
Abstract
AIM:
In the present study, we sought to explore potential differences in the expression and promoter methylation of mitogen-activated protein kinase 1 (MAPK1) between tumor and marginal cells of breast cancer lesions.
METHODS:
A total of 50 randomly selected patients with breast cancer (BCa) undergoing needle biopsy were enrolled. Clinical specimens containing both tumor and marginal cells were collected and preserved. After DNA extraction using specific primers, MAPK1 mRNA and promoter methylation were measured with spectrophotometry at 260/280 nm absorption wavelengths. To deliver a comparative analysis, data from The Cancer Genome Atlas (TCGA) program regarding breast cancer (BRCA), were downloaded from Xena Functional Genomics Explorer and separately analyzed. The suitability of MAPK1 expression and promoter methylation as biomarkers for BCa was analyzed with receiver operating characteristic (ROC) curves.
RESULTS:
We found a positive correlation between tumor stage and MAPK1 expression (P-value: 0.029) in BCa. Likewise, MAPK1 expression was significantly associated with lymph node metastasis (P-value: 0.018). There was a significant difference in the expression of MAPK1 mRNA between tumor and marginal cells of BCa and BRCA (P-value < 0.001). However, we did not find any statistically significant difference in MAPK1 promoter methylation between tumor and marginal cells of both BCa and BRCA. With an area under the curve (AUC) of 0.71, the diagnostic accuracy of MAPK1 expression in BCa and BRCA was validated. However, MAPK1 promoter methylation was not found to be a suitable biomarker.
CONCLUSION:
Our findings suggest that while MAPK1 expression, might be a promising biomarker for evaluating oncogenic activity in patients suspected of BCa. We were not able to detect a prognostic/diagnostic role for MAPK1 promoter methylation.
Introduction
A major epigenetic regulatory mechanism, DNA methylation (DNAm), or promoter methylation in a more specific sense, is a process in which one or more methyl groups are added to the fifth carbon of cytosine residues within certain regions of DNA known as CpG islands (CGIs) [1], which occur near the promoters of about 70% of all mammalian genes [2]. As a general principle, hypermethylation is an indicator of reduced gene expression, whereas hypomethylation is accompanied by euchromatin formation and increased transcriptional activity [1].
While the outstanding frequency of CGIs in our genome indicates the extensive involvement of DNAm in regulation of gene expression in both health and disease conditions, DNAm is of particular importance in the case of neoplasms such as breast cancer (BCa), which had the highest incidence rate of all malignancies among women in 2020, with an estimated number of 2,300,000 new cases [3]. For instance, BRCA1 is a major tumor suppressor gene, the loss-of-function mutations of which are associated with a considerably higher susceptibility to BCa. According to a recently published meta-analysis, promoter methylation of BRCA1 is significantly associated with BCa in Asian populations, i.e., hypermethylation or downregulation of BRCA1 predisposes individuals to BCa [4]. Though, beside BRCA1, several other comparatively important genes have been reported to play pivotal roles in tumorigenesis and patient survival in BCa.
Mitogen-activated Protein Kinase 1 (MAPK1) or Extracellular Signal-Regulated Kinase 2 (ERK2) is a serine-threonine kinase that mediates a critical signal transduction cascade known as RAS-ERK pathway, which is induced in several malignancies including BCa. MAPK1 is involved in tumorigenesis to an extent that a distinct category of anticancer chemotherapeutics termed “ERK2 inhibitors” have been developed for treatment of certain tumors [5]. Some different types of cancers including prostate, liver, renal, and urothelial cancers showed decreased MKP-1 mRNA or protein expression. In 2014, a number of polymorphisms in MAPK1 were linked with increased vulnerability to BCa [6]. As of recent, scientists have revealed that activation of MAPK1 might facilitate resistance of BCa cells to drugs such as tamoxifen [7], bringing about unfavorable clinical outcomes for patients. In addition to this, MAPK1 also stands among several genes that prompt an oncogenic event called epithelial-to-mesenchymal transition (EMT) in normal mammary gland epithelial cells [8], which is highly suggestive of its disease-inducing effects in the margins of BCa tumors that consist of endangered normal cells.
Despite all the information we have about the role of MAPK1 or ERK2 in BCa, few studies have attempted to evaluate the promoter methylation of MAPK1 in the context of BCa to see if there are significant differences in the methylation profile of MAPK1 promoter between tumor and marginal cells in BCa lesions. To this end, we came up with the present investigation to explore the methylation status of MAPK1 promoter and its expression in tumor and marginal BCa cells. In parallel, we also ran a re-analysis of the breast cancer (BRCA) dataset available on The Cancer Genome Atlas (TCGA) Program.
Materials and methods
Investigation of MAPK1 methylation in breast cancer dataset
We assessed MAPK1 gene methylation profile using The Cancer Genome Atlas of the breast cancer (BRCA) (TCGA-BRCA) dataset from high-throughput experiments on breast cancer. TCGA is a public-funded project providing a comprehensive atlas of cancer genomic profiles from large cohorts of more than 30 human malignancies [9]. Methylation beta (β)-values for CpG probes overlapping with MAPK1 promoter on TCGA-BRCA dataset were downloaded using Xena Functional Genomics Explorer (https://xena.ucsc.edu/). Conventionally, β-values approaching a maximum value of 1 indicate hypermethylation, and those nearing 0 denote hypomethylation [10]. For comparison of MAPK1 methylation levels between tumor and marginal tissue samples of breast cancer patients, the mean β-values of all CpG probes were calculated to attain a stable signal, and then analysed. In addition, we analysed the relation between the MAPK1 methylation and expression with the survival of patients using TCGA-BRCA.
Validation of MAPK1 DNAm in tumor and marginal tissue samples of breast cancer patients
Preparation of patient samples
In the present case-control study, tissue samples were collected from 50 patients with breast cancer hospitalized at an institutional tertiary referral hospital within a two-year time interval, spanning from 2017 to 2019. Participants in this investigation were all of Caucasian ethnicity, living in north-western Iran. Patients with a history of radiotherapy or chemotherapy, and those who had refused to participate in this study were excluded. After obtaining written informed consent from all participants, tumor and marginal tissue samples were collected through needle biopsy technique as a routine part of our diagnostic approach. Before genomic DNA extraction, tissues were preserved in liquid nitrogen. The present study was confirmed by the Committee of Ethics in Biomedical Research of Tabriz University of Medical Sciences (ID: IR.TBZMED.REC.1400.064).
List of primer sequences used for determining the expression and methylation levels of MAPK1
List of primer sequences used for determining the expression and methylation levels of MAPK1
The tissue samples were preserved in liquid nitrogen by mortar and pestle, then immediately transferred to extraction kit lysis buffer, followed by homogenization with needle and syringe. Genomic DNA isolation was performed according to the instructions of AllPrep DNA/RNA/Protein Mini Kit (Qiagen, Hilden, Germany). DNA concentration and quality were determined by measuring adsorption at A260/A280 ratio with NanoDrop spectrophotometer (ThermoFisher Scientific Life Sciences, USA). Bisulphite modification was adopted to convert unmethylated cytosine residues (excluding 5-methylcytosine) to uracil, with a mean conversion rate of 99 percent [11].
Methylation-specific qPCR
Methylation of MAPK1 promoter-specific CGIs were examined by quantitative methylation-specific PCR (q-MSP) [12] in the Step One Plus Real-Time PCR System (Applied Biosystems, USA) using BioFACTTM 2X Real-Time PCR Master Mix (Korea). The CGI sequences corresponding to MAPK1 promoters were retrieved from the UCSC genome browser (https://genome.ucsc.edu). The MAPK1 methylation-specific primers were designed with MethPrimer online program (http://www.urogene.org/methprimer). Conditions for amplification were initial denaturation at 95 °C for 15 min, 45 cycles of denaturation for 10s at 95 °C, 30s of annealing at 60 °C and 20s of extension at 72 °C; as the final step, a melting curve analysis was then performed. Primer sequences used for q-MSP are presented in Table 1.
ROC curve analysis
To evaluate MAPK1 promoter methylation as a potential diagnostic biomarker, and investigate its diagnostic accuracy for discriminating patients with breast cancer from healthy individuals, the area under the curve (AUC) of the receiver operating characteristic (ROC) curves was estimated based on DNAm status from TCGA-BRCA cohort and our own clinical samples.
Statistical analysis
GraphPad Prism 6.0 (GraphPad Software, San Diego, CA) was used to perform statistical analyses. To estimate the differences of the p38 MAPK promoter methylation levels between tumor and marginal tissue samples, we used Paired Student’s t-test. To statistically analyse TCGA data, paired and unpaired t-test, Mann-Whitney U test, and Log-rank (Mantel-Cox) test were performed. A p-value < 0.05 was regarded as statistically significant. Data were presented as the mean ± standard error (SD) of the experiments.
Results
The clinicopathological of specimens is listed in Table 2, which also provides a statistical overview of correlations between each clinical feature and MAPK1 expression/methylation. A total of 50 samples, containing tumor and marginal cells, were collected from 50 female patients with a mean age of 56.7, the majority of whom were older than 55. While there was no statistically significant correlation between age and MAPK1 expression/methylation, we noted a positive association between tumor stage and MAPK1 expression, as samples obtained from patients with stage IV BCa (n = 7, 14%) displayed the highest MAPK1 expression. While a slightly significant association was also noted between tumor stage and MAPK1 methylation, a p-value > 0.05 rendered this observation insignificant.
Clinicopathological features of patients with breast cancer and their correlation with MAPK1 expression and methylation
Clinicopathological features of patients with breast cancer and their correlation with MAPK1 expression and methylation
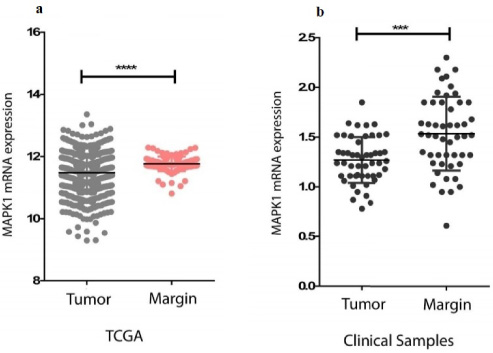
MAPK1 mRNA expression. (a) Expression level of MAPK1 mRNA in tumor and marginal cells of breast cancer based on data adapted from TCGA. (b) Comparison of MAPK1 mRNA expression in tumor and marginal cells of clinical specimens collected from BCa patients. Asterisks indicate statistical significance, as *** denotes a p-value < 0.001 and **** denotes a p-value < 0.0001.
Lymph node metastasis (LNM) was present in 29 (58%) patients, and exhibited a positive correlation of statistical significance with MAPK1 expression. Though, no association was found to exist between LNM and MAPK1 methylation. Similarly, a history of chemotherapy was not found to affect MAPK1 expression/methylation to a significant extent (Table 2).
We analysed MAPK1 promotor methylation in tumor and marginal tissue samples collected from patients with breast cancer using clinical samples and the data available on TCGA-BRCA. The pattern of MAPK1 promotor methylation in different samples showed that the methylation rate in tumor cells was slightly higher than the marginal cells, though, there was not a significant relationship between methylation and the spatial occurrence of tumor cells (Fig. 1a). This was in agreement with TCGA-BRCA, according to which there was no remarkable difference between MAPK1 promotor methylation rates in BCa tumor and marginal tissues (Fig. 1b).
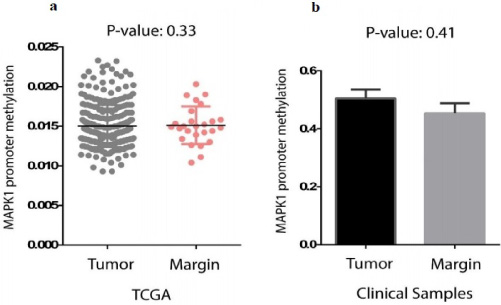
MAPK1 promoter methylation. (a) Promoter methylation of MAPK1 in tumor and marginal cells of breast cancer on data adapted from TCGA, indicating insignificant difference between the two cell types with a p-value of 0.33. (b) Promoter methylation of MAPK1 in tumor and marginal cells of clinical specimens collected from BCa patients. A p-value of 0.41 indicates that the pattern of promoter methylation between tumor and marginal cells was not statistically different.
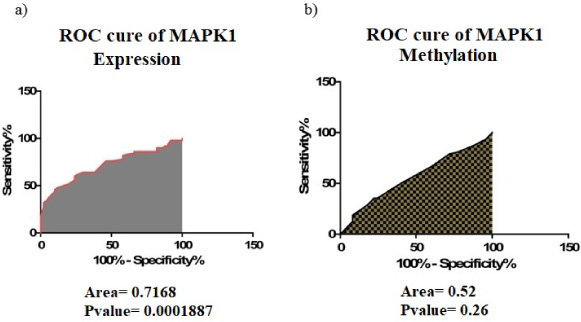
ROC curve of MAPK1 expression (a) and methylation (b) based on data adapted from BCa clinical specimens, suggesting that the expression level of MAPK1 might be of value as a biomarker.
We investigated the expression of MAPK1 in tumoral and marginal regions of breast cancer, and found that MAPK1 was significantly upregulated in marginal regions compared with tumor tissues in both clinical specimens (Fig. 2a) and TCGA dataset (Fig. 2b). Thus, we concluded that there was a significant difference between the expression of MAPK1 in tumoral and marginal samples. However, this difference was not due to differential methylation of MAPK1 between the two tissue types, as there was not a statistically significant correlation between promoter methylation of MAPK1 and the location from which clinical specimens were obtained.
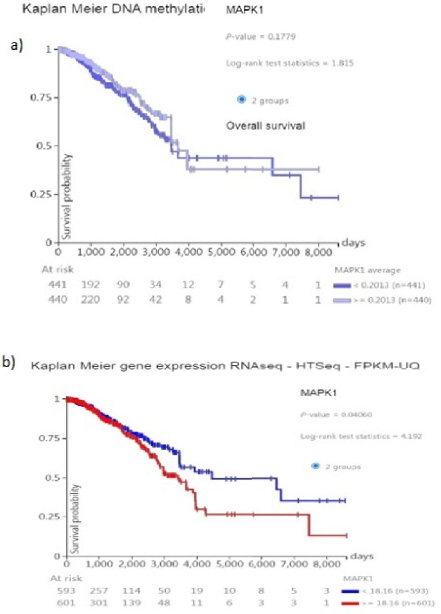
(a) Correlation between Promoter methylation of MAPK1 in tumor cells of breast cancer on data adapted from TCGA and survival rate of patients. (b) Correlation between expression of MAPK1 in tumor cells of breast cancer on data adapted from TCGA and survival rate of Patients (Kaplan Meier plot analyses).
We adopted ROC curves to examine the value of MAPK1, in terms of methylation (Fig. 3a) and expression (Fig. 3b), as a biomarker for diagnosis of BCa, and found that the expression of MAPK1 in 72.68% of marginal samples was actually higher than tumor tissues. In this sense, the expression level of this gene can be used as a diagnostic biomarker in patients suspected to have BCa. ROC curves for MAPK1 promoter methylation indicated that in only 52% of the marginal tissues the rate of methylation was higher than tumor tissues, but this difference in methylation was not sufficient to be recognized as a diagnostic biomarker. Thus, the expression level of MAPK1 could be considered a diagnostic biomarker in the case of BCa, however, the methylation profile of this gene confers negligible, if any, value in terms of BCa diagnosis.
MAPK1 expression and patient survival
Because we don’t have access to the survival rate of our patients Kaplan-Meier diagram of TCGA data set used to identify the relation between the survival rate of the patient with breast cancer and MAPK1 promoter methylation (Fig. 4a) and expression (Fig. 4b) with patient survival, we found that MAPK1 methylation in BCa was not significantly correlated with patient survival (p-value:0.1779). However, MAPK1 expression, regardless of its promoter methylation, was shown to have a significant correlation with patient survival (p-value: 0.04).
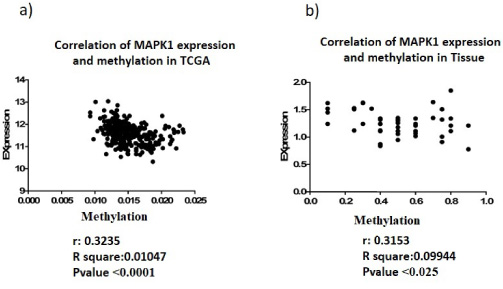
(a) Correlation between Promoter methylation of MAPK1 and expression level of in tumor cells of breast cancer on data adapted from TCGA. (b) Correlation between Promoter methylation of MAPK1 and expression level of in tumor cells of breast cancer on tissue samples.
Correlation analyses between MAPK1 methylation and expression confirm the significant negative correlation between the promoter methylation and expression level of this gene, both in tissue samples (r: −0.3153, p-value: 0.025) (Fig. 5a) and TCGA data set (r: −0.3235, p-value < 0.0001) (Fig. 5b).
Discussion
It has long been known that the marginal tissue in BCa patients is of significant predictive value in terms of survival. For instance, patients undergoing breast surgery with tumor margin excision were four times less likely to develop recurrent BCa, compared with those patients who had undergone less extensive surgery with partially excised or intact margins [13]. These findings indicated that the marginal tumor cells in BCa possess certain transcriptional features that renders them capable of determining the prospective progress of the disease. Consistent with these findings, our investigation demonstrated that there is, in fact, a significant relationship between the expression of marginally-upregulated MAPK1 and patient survival, noting the influential role of marginal cells in the pathogenesis of BCa.
As of late 2021, an investigation by Hamadneh et al. on tamoxifen-resistant BCa cells revealed that upregulation of MAPK1 was associated with higher incidence of resistance to tamoxifen, predicting a poor clinical outcome with regards to increased likelihood of metastasis [14]. Prior to this, Rizeq et al. had reported that inhibition of MAPK1 by means of Chalcone analogues promoted apoptosis in BCa cells [15], confirming the findings disclosed earlier that year by Kheraldine et al. who had successfully induced apoptosis in BCa cells by repressing MAPK1 through administration of poly(amidoamine) dendrimers [16]. Another important discovery in 2021, regarding the role of MAPK1 in BCa, was made by Deng et al. who suggested that MAPK1 was responsible for reduced rate of mitophagy, a mitochondrion-based form of autophagy, in BCa and migration of metastatic cells to bones [17]. Consistent with all these investigations, we observed increased expression of MAPK1 in marginal tissues, and found that this could have a significant impact on patient survival, confirming a pro-metastatic role for MAPK1.
In 2020, Gagliardi et al. documented the positive regulatory effect of MAPK1 on development of cancer stem cell (CSC) phenotype in triple-negative BCa once they observed that MAPK1 knockdown would result in decreased self-renewal capacity of BCa cells [18]. The MAPK pathway is often found to be highly activated in TNBC. TNBC is associated with a younger age at diagnosis, advanced stage at diagnosis, increased risk of visceral metastasis, and poorer outcome. These are not responsive to conventional receptor-target therapies also [19]. Earlier that year, Castillo-Sanchez et al. found out that MAPK1 might mediate the oncogenic effects of the toxic compound Bisphenol A on migration of BCa cells [20], providing further grounds for the observations made in the present work.
Four years earlier, in 2016, Rincón et al. showed that overexpression of MAPK1 was associated with resistance to taxanes, e.g., docetaxel and paclitaxel, tumor relapse and decreased disease-free survival [21]. These findings were highly consistent with those of another study, in which Smith et al. used a specific small interfering RNA (siRNA) to silence MAPK1 in BCa cells. They found that MAPK1 gene silencing reverted epithelial-to-mesenchymal transition (EMT) in normal mammary gland epithelial cells, indicating the oncogenic nature of MAPK1 [22]. In line with these reports, we identified significantly increased expression of MAPK1 in marginal tissues that might very well be an indicator of tumor progression.
In contrast to its expression, MAPK1 has less frequently, if any, been investigated for its methylation profile in BCa. In fact, to date, only one study has evaluated the promoter methylation of a MAPK-related gene in BCa, which was reported in 2012 by Chen et al. [23], who measured the expression of MAPK phosphatase-1 (MKP-1) in five different cell lines of BCa (MCF7, T47D, MDA-MB-231, SKBR3, and BT474), and compared the results with that of normal mammary gland cells (M10). As the name suggests, MKP-1 is an enzyme that catalyzes dephosphorylation and inactivation of MAPKs. This study showed that all of the five BCa cell lines had significantly reduced expression of MKP-1, indicating higher activity of MAPKs and potentially MAPK1. Consistently, Chen et al. also discovered that the MKP-1 promoter was significantly hypermethylated in BCa cells when compared to the normal cells, which showed almost no methylation, highlighting lower expression of MKP-1 and higher activity of MAPKs in malignant cells. As such, they argued that the promoter methylation of MKP-1 might be a candidate biomarker for predicting pro-malignant activity in BCa.
In the other part of our study we confirm the negative correlation between the methylation and expression level of MAPK-1 in tumor samples which showed the importance of the identification of genes that change the promoter methylation status might help to discover new biomarkers for tumor diagnosis and prognosis for example, hyper methylation of RASSF1A, HIN-1, RAR-β, Cyclin D2, and Twist genes has been reported in early diagnosis for breast cancer [24].
Regarding to our data, we confirm a negative but weak correlation between the promoter methylation and MAPK-1 expression. This supposed that in addition to methylation, other factors such as post-transcriptional regulation by non-coding RNAs can play an important role in regulating of this gene. In addition due to our results MAPK-1 methylation is not as suitable biomarker for breast cancer.
Though we were not able to detect a prognostic/diagnostic role for MAPK1 promoter methylation, in contrast to that of MKP-1, we still revealed a significant correlation between MAPK1 expression and patient survival, indicating the oncogenic effect of MAPK1 in marginal cells of BCa tumors. Our observations further extend the scope of the earlier works, especially the Chen et al. study, while highlighting the potential prognostic value of MAPK1 levels, but disputing that of MAPK1 promoter methylation, in BCa.
Footnotes
Acknowledgements
None.
Ethical statement
With permission from the Committee of Ethics in Biomedical Research of Tabriz University of Medical Sciences, tissue samples were obtained from the Department of Oncology and Hematology, Faculty of Medicine, Tabriz University of Medical Sciences (approval ID. IR.TBZMED.REC.1400.064).
Conflict of interest
None.
Data availability statement
Funding
The present investigation was funded by Tabriz University of Medical Sciences (Grant ID: IR.TBZMED.REC.1400.064).
Author contributions
SG and MR designed the study. MA performed the laboratory experiments. MR and HZ supervised the study. SG and MS wrote the first draft of the manuscript. MS edited the manuscript. All authors have read and approved the final version of the manuscript.