
Abstract
BACKGROUND:
Breast cancer, one of the leading causes of cancer-related mortality in women worldwide, exhibits wide-ranging histo-morphologic, clinical and molecular diversity.
OBJECTIVE:
This study compares the genetic alterations of breast tumors with the histo-morphological, hormone receptor status and metastatic “organotropism”.
MATERIALS AND METHODS:
Twenty-two cases of primary invasive breast carcinoma with local/distant metastasis were retrieved from the pathology archives. The status of estrogen and progesterone receptors by immunohistochemistry was recorded along with other pertinent case data. Next generation sequencing was performed on formalin-fixed paraffin embedded blocks of tumor.
RESULTS:
The mean age of the study subjects was 57.9 ± 13.3 years. TP53 mutation was the most common gene alteration in this study and was seen in 40.9% cases. ROS1 gene was mutated in 44.4% PR negative breast cancers while being wild type in the twelve PR positive tumors. (p = 0.021).
STRING interaction network constructed with ROS1 and PR revealed a significantly higher number of interactions in this network than expected (p-value 0.000973).
CONCLUSION:
This study highlights the significantly higher incidence of ROS1 gene alterations in metastatic PR− breast cancers, with STRING network analysis revealing higher nodal interaction in the nodal network comprised of PR and ROS1 exclusive of ER.
Introduction
Breast cancer is the most common cancer in women and one of the leading causes of cancer-related mortality in women world wide [1]. Breast cancer exhibits wide-ranging histo-morphologic, clinical and molecular diversity [2,3]. This necessitates the use of multiple means to classify these tumors.
Based purely on morphological features, breast cancers are classified into ductal, lobular and other special types [4]. Histological typing, as per College of American Pathologist’s consensus statement, is a category I prognostic factor for breast cancers. Despite the lack of prognostic implications in certain special types, their recognition is essential to prevent diagnostic conundrum and errors [4].
Based on hormone-estrogen (ER) and progesterone (PR)-receptor status, breast cancers can be classified into four distinct subgroups, ER+PR+, ER+PR−, ER−PR+ and ER−PR− [5,6]. The gene expression of PR is estrogen dependent and PR expression is contingent on intact ER response pathway [7]. Consequently, the ER−PR+ group is widely accepted to be a technical artefact due to suboptimal tissue fixation or low sensitivity assays [8]. Though PR expression is a strong prognostic factor in ER+ breast cancer, ambiguity persists regarding the independent role of PR in breast cancers.
Extensive studies to decipher the genomic intricacies of invasive breast carcinoma led to the identification of four subtypes- luminal A, luminal B, Her 2- positive and basal-like [9]. This molecular classification is based on the molecular expression profiles of biomarkers including hormone receptors and human epidermal growth factor 2 (HER2), which are routinely assessed by immunohistochemistry (IHC) [10]. However, this classification is by no means all-encompassing and several gaps remain in our understanding of the genomic complexity of breast cancer [11–15].
Lastly, distant metastases are the cause for 90% cases of mortality in breast cancer. Distant metastases to certain organs is a non-random, complex process, referred to as metastatic “organotropism”, determined by various tumor and host factors, and crosstalk [16]. Genetic alterations in both the tumor cells and the target organ play a synchronized role in this metastatic organotropism and unravelling these molecular pathways may pave the way for novel therapeutic stategies [17].
It is essential to elucidate these elusive molecular pathways in order to, not only, improve the understanding of disease prognosis but also expand the therapeutic modalities and refine targeted therapy. This study compares the genetic alterations of breast tumors identified by tumor sequencing with the histo-morphological, hormone receptor status and metastatic “organotropism”, in an attempt to identify and clarify the genetic pathways contributing to the intricacies of this multifaceted disease.
Materials and methods
The study was approved by the Institutional Review Board and Ethics Committee of Mount Sinai Medical Center, Miami Beach, Florida.
Case selection
Twenty-two cases of primary invasive breast carcinoma with local/distant metastasis were retrieved from the pathology archives. Cases with other prior or concomitant malignant neoplasms were not included in this study. The status of estrogen and progesterone receptors in the tumor by immunohistochemistry (IHC) was recorded along with other pertinent case data including stage, grade, histological type, presence and location of metastasis, HER2/neu status as well as survival data.
Genetic testing and data analysis
Formalin-fixed paraffin embedded tissue blocks, with adequate quantity of viable tumor, were selected and sent out for next-generation sequencing. A panel of 324 genes were examined for genetic alterations at an independent laboratory. The data obtained from sequencing these cases were analyzed for recurrent genetic abnormalities in an attempt to delineate phenotypic and prognostic associations. Statistical analysis was performed using IBM SPSS 26 software (Armonk, NY, USA). Nonparametric data was assessed for statistical significance using Fisher exact and Chi square tests.
ROS1 mutation and PR expression by IHC were mapped to the search tool for retrieval of interacting genes (STRING) to acquire protein–protein interaction (PPI) networks. The STRING tool was employed to seek potential interactions between ROS1 and PR expression using a full STRING network and physical subnetwork. Cytoscape software version 3.6.1 was used to visualize the PPI network. Active interaction sources, including text mining, experiments, databases, co-expression, neighborhood, gene fusion and co-occurrence, and a minimum interaction score > 0.4 were applied to construct the PPI networks. The species limited to “Homo sapiens” and the whole genome was the assumed statistical background for the enrichment analysis.
Results
A total of twenty-two cases of metastatic breast carcinoma in women were analyzed in this study. Based on histology, eighteen cases were classified as invasive ductal carcinoma, no special type. There were two cases of invasive lobular carcinoma while one case had features of both invasive lobular and invasive ductal carcinoma and one case was classified as metaplastic carcinoma.
The mean age of the study subjects was 57.9 ± 13.3 years. Twelve cases were ER/PR positive, while four were ER positive but PR negative. The ER/PR status was unconfirmed in one case. All but one (Stage 2) of the cases were clinically stage 4, at the time of sequencing, with distant metastasis to one or more sites. Liver metastases were present in eleven cases, bone metastases in twelve cases, lung and brain metastases were seen in four cases each, while skin metastases were present in two cases and one case had metastasis to the chest wall. Nine cases had metastasis to multiple sites. At the time of data collection, 7 patients were deceased while the remainder were under follow-up. Characteristics of the study subjects is summarized in Table 1.
Characteristics of study subjects
Characteristics of study subjects
∗Data not available for one case.
Of the 324 genes analyzed by sequencing, 144 were altered in the 22 cases included in this study (summarized in Table 2).
Genes altered in 22 cases of invasive breast carcinoma
TP53 mutation was present in 9 cases (40.9%) and was the most common gene alteration in this study. The other frequently altered genes were CCND1, FGF19 and FGF3, which were seen in eight cases (36.4%) each, while FGF 4 alterations were present in 7 cases (31.8%).
The genetic alterations were seen in an average of 17 genes in cases with metastasis to multiple sites as compared to 11 genes in cases with single site metastasis (p > 0.005). No particular gene alteration was found to show a statistical association with the number or site of metastasis.
ROS1 gene was mutated in four of nine (44.4%) PR negative breast cancers while being wild type in the twelve PR positive tumors (19% of all cases). This association was significant with a Fisher-exact p value of 0.021. No such association was seen with the ER status which was positive in two of the ROS1 mutated PR negative cases and negative in the other two.
STRING interaction network constructed with ROS1 and PR revealed there were a total of twelve nodes with 38 edges (interactions) in this network as opposed to the expected number 22 (Fig. 1). There was significantly higher number of interactions in this network than expected. The PPI enrichment p-value was 0.000973, indicating that the proteins are at least partially connected in biological functionality. The average node degree was 6.33 and the average local clustering coefficient was 0.747. The predicted functional partner score was greater than 0.9 between the various nodes (Fig. 2).
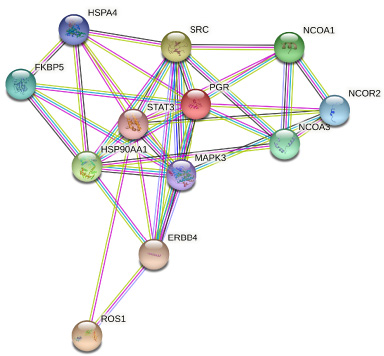
STRING interaction network constructed with ROS1 and PR with twelve nodes and 38 edges (interactions).
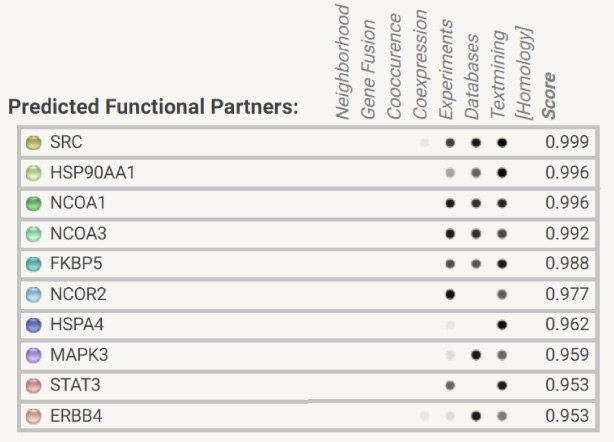
Predicted functional partner score between the various nodes.
The clinical heterogeneity of breast cancer has been long explored in an attempt to refine risk prognostication and management strategies. Rapid progression and expansion of high throughput sequencing and bioinformatics have uncovered the comprehensive molecular heterogeneity of breast cancer [2,3].
TP53 is the most commonly mutated gene in breast cancers across multiple studies and is implicated in the initiation and progression of 20–40% breast cancers [18,19]. P53, the tumor suppressor protein encoded by the TP53 gene, when mutated promotes mammary carcinogenesis of the stromal type. TP53 mutations triggers breast cancer in middle aged women at frequency comparable to BRCA1. The reported frequency of TP53 mutations in metastatic breast cancer varies from 40% to greater than 70% across various study cohorts [18].
Our study cohort reflected the lower end of this spectrum with close to 41% cases harboring mutations in TP53. Other commonly mutated genes in our cohort included CCND1, FGF3 and FGF19 which have been commonly reported in breast cancers with similar frequency as seen in our study group [18].
With reference to “organotropism” of metastasis, the study failed to identify any genetic alteration showing statistically significant association with metastasis to a particular site or organ. This could be attributed to multiple factors including the small study population and the widely metastatic nature of tumor in most of the study populations contributing to the confounding effect. Identification of molecular basis of “metastatic organotropism”, would require a much larger study set with single organ metastasis which is beyond the scope of our study.
Intrinsic molecular heterogeneity results in variable response to different treatment modalities, necessitating elucidation of molecular disposition of tumors for optimal risk stratification and treatment [20–22]. With the advent of targeted therapy, identifying oncogenic drivers that are targetable can vastly improve prognosis of breast cancer patients [2,11,23–25]. Exposition of analogous and surrogate IHC markers that can help in molecular stratification of cases will further improve the efficacy and impact of genomic medicine. The molecular expression-based classification of breast cancer into Luminal-A, Luminal-B, HER2–positive and basal-like is one such risk prognostication strategy that can be evaluated via IHC analysis of analogous markers ER, PR Her2 and Ki67. However, this classification is not all encompassing and needs to be further expounded [26].
For instance, higher PR is seen more frequently in luminal A tumors (favorable outcome) as compared to luminal B (worse prognosis) [27]. ER+PR− tumors are substantially more aggressive than ER+PR+ tumors, with higher mortality [28,29]. The molecular contrivance of this phenomenon remains elusive. The lack of PR expression in ER+ tumors was attributed to decreased ER activity and considered predictive of limited endocrine responsiveness [30]. However, this theory fails to explain the sensitivity of some ER+PR− tumor to endocrine therapy [31,32]. ER+PR− tumors are predominantly HER2 negative and the role of PIK3-Akt-mTOR signalling due to HER2 amplification remains dubious in their etiology [33–35]. Thus, the ambiguity surrounding the etiopathogesis of ER+PR− tumors remains.
ROS1 gene, located on chromosome 6q22.1, encodes a receptor tyrosine kinase (RTK) that regulates morphogenesis and cell differentiation in several organs [36,37]. ROS1 alterations including gene fusions have been described in breast cancers [38]. ROS1 gene alterations were found to be exclusive to the PR− subset of metastatic breast cancers in our study. Stein et al. [39] in their study of 78 breast cancer patients found ROS1 mutations to be the most common variant of unknown significance (VUS) of 29 genes encoding cancer implicated RTKs in their patient database. Most of the mutations were predicted to be damaging. Their reported ROS1 mutational frequency of 21% is close to the 19% found in our study.
The striking difference though, is the exclusive localization of ROS1 mutations to the PR− subset of metastatic breast cancers included in our study. These ROS1 mutated cases may potentially represent a distinctive subtype of PR− breast cancers. This statistically significant association was further explored with the help of a STRING network constructed using Cytoscape [40]. The STRING network revealed increased statistically significant network interactions and strong functional correlation. Though the number of cases included in this study is low, the findings achieved statistical significance. Nearly 50% PR− breast cancers included in the study showed ROS1 alterations. Such gene alterations such as ROS1 in proteins related to the PR nodal network and neighbourhood not including ER as demonstrated in the STRING network, provide pathways that explain the independent role of PR repression in breast cancer in tumor biology influencing aggression and prognosis.
ROS1 gene alterations including somatic chromosome fusions have been reported in a wide variety of cancers in both the adult and pediatric population [41]. The altered kinase domain of ROS1 becomes constitutively active, driving proliferation of tumor cells [42]. The advent of tyrosine kinase inhibitors (TKI) has allowed targeted treatment of kinase domain activated cancers. ROS1 gene shows structural homology to ALK and this was the original basis of a trial of ALK inhibitors in ROS1+ lung cancers leading to a remarkable progression free survival of 19.2 months with crizotinib in PROFILE 1001 trials [43]. This led to FDA approval of crizotinib as first line therapy in ROS1+ lung cancer and exploration of TKIs as a possible therapeutic option in other ROS1 altered tumors. Trials of crizotinib in ROS1 fusion breast cancer have shown response to treatment [44,45]. As such, TKIs targeting ROS1 may be an alternative modality for escalated therapy in PR− breast cancers.
Though the number of cases included in the study is small, the statistically significant finding of ROS1 mutations in a subset of PR− metastatic breast cancers, may hold the key to understanding the biology and behaviour of PR− breast tumors, as well as identifying novel therapeutic strategies such as TKIs in these tumors. Thus, this finding merits further evaluation in a larger cohort of patients with metastatic breast cancers.
Conclusion
This study highlights the significantly higher incidence of ROS1 gene alterations in metastatic PR− breast cancers, with STRING network analysis revealing higher nodal interaction in the nodal network comprised of PR and ROS1 exclusive of ER.
Footnotes
Conflict of interest
The authors certify that they have no affiliations with or involvement in any organization or entity with any financial or non-financial interest in the subject matter or materials discussed in this manuscript.