
Abstract
Alzheimer’s disease (AD) is the most common type of dementia, which is characterised by progressive memory loss and accumulation of hallmark markers amyloid-β (Aβ) and neurofibrillary tangles in the diseased brain. The current gold standard diagnostic methods have limitations of being invasive, costly, and not easily accessible. Thus, there is a need for new avenues, such as imaging the retina for early AD diagnosis. Sleep disruption is symptomatically frequent across preclinical and AD subjects. As circadian activity, such as the sleep-wake cycle, is linked to the retina, analysis of their association may be useful additions for achieving predictive AD diagnosis. In this narrative review, we provide an overview of human retina studies concerning the deposition of Aβ, the role of the retina in sleep-wake cycle, the disruption of sleep in AD, and to gather evidence for the associations between Aβ, the retina, and sleep. Understanding the mechanisms behind the associations between Aβ, retina, and sleep could assist in the interpretation of retinal changes accurately in AD.
INTRODUCTION
Dementia is categorized as the second most leading cause of death in Australia [1]. Alzheimer’s disease (AD) is the most common type of dementia and an estimated 401,300 Australians are currently living with dementia [1]. There are currently little to no curative therapies for AD; therefore, prevention through managing modifiable risk factors is a prioritized endeavor [2]. Lifestyle interventions provide a promising evidence-based AD prevention strategy, yet the optimal time for administering these interventions in the disease trajectory is still uncertain [3–7]. Although prevention hinges on the early diagnosis of the disease, definitive diagnosis of AD is only possible when the primary pathological features of AD, namely plaques comprising of amyloid-β (Aβ) protein and neurofibrillary tangles comprising of tau protein, are identified during autopsy of the brain [8]. However, there is now evidence that AD related Aβ pathogenesis in the brain can be witnessed 10–15 years before clinical symptoms are apparent [9].
The current gold standard for AD diagnosis includes measuring the Aβ load in the brain using positron emission tomography (PET) or measuring both Aβ and tau protein levels in the cerebrospinal fluid (CSF) [9]. These tools are recommended for research purposes only and have caveats in being highly invasive, expensive, and not widely available for use in a clinical setting [10]. The retina is the neurosensory structure of the eye and is a direct extension of the central nervous system as it is derived from the same embryological tissue as the brain [11]. Therefore, in the past few years, novel diagnostic avenues such as imaging the eye have gained significant attention to identify AD related biomarkers which could reflect the build-up of Aβ in the brain.
In vivo studies document AD related changes in the retina, including a decreased thickness of the retinal nerve fiber layer (RNFL), changes in the retinal blood vasculature, and identification of retinal Aβ in the postmortem transgenic mice and human AD retina [12–14]. However, studies using retinal imaging to identify Aβ in the live human retina have provided inconclusive results [13, 16].
Therefore, a more comprehensive tool that incorporates surrogate information could be more useful to endorse AD specific retinal changes. Indeed, studies have identified the role of the retina in sleep and circadian rhythm (sleep-wake) disruption in AD [17–19]. Here, we review the crosstalk between the retina, Aβ, and sleep-wake systems to evaluate sleep-wake disturbances as surrogate markers in future retinal imaging and early AD diagnostic studies.
METHODS
An extensive literature search was conducted across PubMed and Google Scholar databases to include relevant papers pertaining to studies correlating aspects of the relationship between Aβ, retina, and sleep-wake systems. Figure 1 displays the flow of studies included in this review. Briefly, information regarding sleep, dementia, and the retina was first obtained across Google scholar databases. Secondly, inclusive of these search terms, a combination of keywords was applied in PubMed database: Aβ, retinal Aβ, circadian rhythm, Alzheimer’s disease, dementia, sleep, rest-wake cycle, orexin, ghrelin, leptin, and cognition for scientific literature conducting human or animal observational, interventional, or review studies. Titles and abstract screening were conducted to preliminary, or unrelated studies. Exclusion criteria (see Fig. 1) were reviewed manually and applied to the eligible papers. This search was performed and updated through August 2020 – December 2023.
Flowchart of studies included.
Eligible studies were published in English, peer reviewed clinical, observational, interventional or review studies. Each paper evaluated or measured a variable concerning the retina, Aβ and sleep-wake functions on the sample cohort or in the reviewed literature. For the clinical, observational, and interventional studies, the sample inclusion criteria required participants who were aged≥50 years, with an assessment of cognitive functions measured. Publications excluded from the extracted data include: 1) studies of populations with ophthalmological or retinal diseases, 2) articles measuring vasculature or including angiography as the only variable, 3) studies without retinal measures, and 4) review papers.
Two reviewers (ID and TS) independently sourced publications according to the inclusion criteria. Eligible articles were then identified for the planned narrative review. Disagreements between the reviewers about study inclusion were resolved through discussion. Specifically, the data extracted (see Table 1) includes sample size, clinical diagnosis, age, study technique/intervention, and a summary of the main findings.
Evidence supporting the relationships between sleep, the retina, and amyloid-β in AD
AD, Alzheimer’s disease; Aβ, amyloid-beta; IHC, immunohistochemistry; MMSE, Mini-Mental State Examination; HIS, hyperspectral retinal imaging; PET, positron emission tomography; MCI, mild cognitive impairment; FAF, fundus auto fluorescence; OCT, optical coherence tomography; mRGC, melanopsin retinal ganglion cell; PLR, pupillary light reflex; RCT, randomized control trial; CN, cognitively normal; CSF, cerebrospinal fluid; ELISA, enzyme-linked immunosorbent assay; PSQI, Pittsburgh Sleep Quality Index; MRI, magnetic resonance imaging; p-tau, phosphorylated-tau; OSA, obstructive sleep apnea; RNFL, retinal nerve fiber layer; APOE, apolipoprotein E.
Aβ In The Human Retina
Studies utilizing postmortem transgenic AD mice retina and human AD retina have detected Aβ and tau pathology, within various tissues of the eye, including the lens, vitreous humor, and the retina [20, 21]. Aβ is reported deposited earlier in the retina than the occurrence of cognitive deficits [14, 22]. Particularly, the pioneering study by Koronyo et al. used Aβ specific curcumin (L. Curcuma longa) staining in both postmortem AD human and AD mice retina, reporting that Aβ like plaques were deposited in the retinal ganglion cell (RGC) and RNFL layers, and vasculature in vivo. In 20 participants, with mild cognitive impairment (MCI; a high risk state for AD) and 20 healthy controls aged 66–84 years, Kayabasi et al. used fundus autofluorescence to detect lesions of interest and optical coherence tomography (OCT) to determine the retinal layer of Aβ deposition [23]. Identified exclusively in the MCI group, curcumin staining’s affinity for Aβ increased the fluorescence of abnormal deposits within the outer plexiform, RGC and RNFL layers surrounding the macula regions and retinal vasculature [23]. However, these studies did not compare correlative brain PET Aβ imaging which could demonstrate the association of deposits within the retina to brain Aβ load. On the other hand, Snyder et al. in a cross-sectional study of 63 cognitively normal adults with a family history of AD, investigated the comparison between brain Aβ load, and retinal biomarkers identified with spectral domain OCT [24]. Deposits termed as ‘inclusion bodies’ size and retinal layer volume was significantly different in the inner plexiform layer (IPL) and this relationship of deposit size and retinal layer volume increased with brain Aβ load [24]. However, the major limitation to this study is the lack of conclusive post-mortem analysis of the inclusion bodies containing Aβ.
In a review report, Shah et al. noted that retinal Aβ may be present in other conditions; for instance, drusen found in macular degeneration [9]. Therefore, it is important to determine whether Aβ deposition in the retina can be considered as a specific signature AD marker. Furthermore, Shah et al. noted that hyperspectral imaging (HSI) may be a more sensitive method in identifying retinal Aβ and assist in the differential diagnosis from other conditions. The main advantage of HSI appears able to detect AD related retinal fluorophores without the need for external labelling, as shown in Fig. 2. HSI also addresses the limitation of non-specific staining by curcumin, as it is not an antibody. Studies to identify retinal Aβ and associated changes in the human retinas are currently underway using this method. Sharafi et al. reported that retinal vasculature and texture changes could discriminate PET Aβ positive from Aβ negative subjects, with 85% accuracy, using HSI [13]. Hadoux et al. reported that the retinal reflectance spectra measured using HSI could differentiate PET Aβ positive MCI subjects from PET Aβ negative controls [15]. More et al. have used HSI in a cohort of 19 AD participants and 16 healthy controls grouped on the basis of their Mini-Mental State Examination (MMSE) scores. Their findings indicated the specific spectral signatures generated by the interaction of light and reflective structures in the retina such as, layers of the retina, blood supply, and AD-related Aβ deposits, are feasible in detecting early stages of the disease, i.e., MMSE score≥22 [22]. However, studies using retinal imaging to identify Aβ in the live human retina have provided inconclusive results as compared with the results of Sharafi et al. and Hadoux et al. only reporting correlative outcomes as indirect measures of retinal Aβ [13, 16].
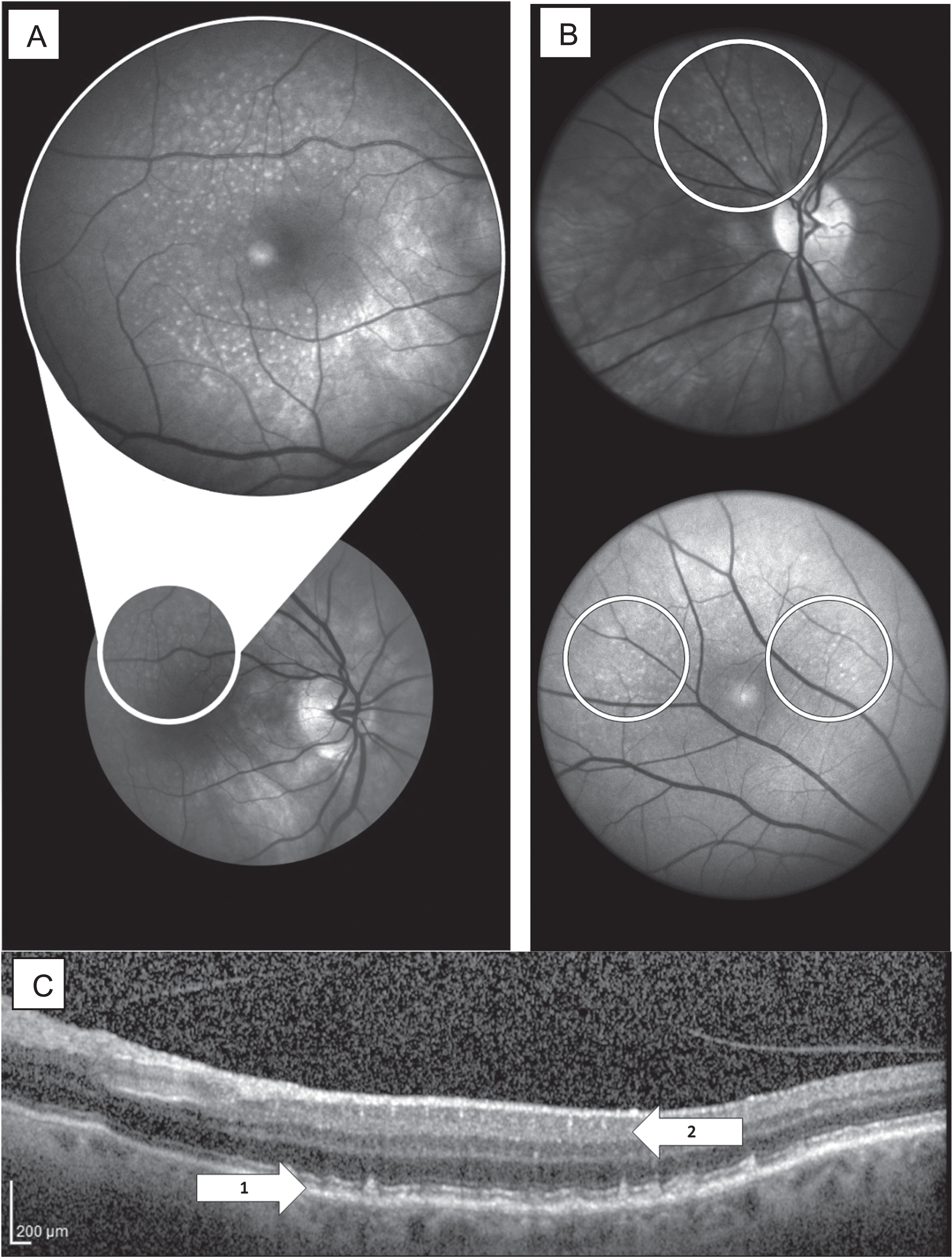
Presentation of deposits in retinal imaging. A) HSI of the retina at 575 nm displaying drusen deposits in the macular region. B) Examples of HSI between 450–580 nm showing deposits of interest outside of the macular region. C) OCT image illustrating drusen deposition in and between the RPE and Bruch’s membrane retinal layers (1). Note: According to literature, the layers of deposition for Aβ are in superior layers, including the RNFL, GCL, and IPL (2) [14], [54]. HSI, hyperspectral imaging; OCT, optical coherence tomography; RPE, retinal pigment epithelium; RNFL, retinal nerve fiber layer; GCL, ganglion cell layer; IPL, inner plexiform layer.
Measuring sleep-wake activity and considering its use as a biomarker is plausible due to the incidence of specific patterns seen in oscillatory events, such as slow wave activity, spindle density, and slow oscillation-sleep spindle, being associated with the presence of AD neuropathology [25]. In support, Naismith et al. identify that MCI individuals present with circadian misalignment and sleep disruption caused by changes in secretion of the key sleep hormone melatonin, which is consistent with changes seen in AD [26]. Therefore, analyzing additional symptomatic information, such as sleep-wake functions, could help explain associated changes occurring in the retina. Combined use of surrogate markers may be another way to conclusively determine whether changes in the retina can reflect the buildup of Aβ in the brain in AD.
EYEING ON SLEEP
The circadian rhythm-sleep-retina link
The human body’s internal clock or circadian rhythm is a 24-h cycle programmed to carry out essential functions and processes. The sleep-wake cycle, otherwise termed rest-activity cycle is one of the most important and well-studied circadian rhythms. In healthy individuals, the timing of sleep is regulated by the circadian rhythm and is directly influenced by environmental cues, including light [27]. The circadian rhythm (including the sleep-wake cycle) is synchronized with the central pacemaker, the suprachiasmatic nucleus (SCN), located in the brain’s hypothalamus. It regulates the key circadian markers, namely melatonin and cortisol. These markers, in turn, govern the fluctuations (rise and fall) of core body temperature which are closely tied with sleep-wake timing [27]. Melatonin is a hormone known for its function in inducing sleep and is mainly produced in the pineal gland [19]. Cortisol is a hormone known for its function in response to stress, though it also plays a role in inducing wakefulness, and is produced in the adrenal glands [28]. Under physiological conditions, the body temperature drops, cortisol levels decrease, and melatonin levels increase to induce nocturnal sleep, and vice versa for wakefulness during the day [27]. This intrinsic system can be entrained with external cues such as the light/dark cycle, temperature, or physical activity [27].
Literature shows that the neuronal retina plays a role in stimulating the expression of melatonin and therefore contributes to sleep induction [19]. Normally, light activates melanopsin in the retinal ganglion cells (mRGCs) of the inner retina and starts a cascade of events which results in an increased expression of melatonin. mRGCs are intrinsically photosensitive cells and are responsible for the non-image forming functions, including detection of contrast, a set of light responses that include circadian entrainment, pupillary light reflex (PLR), and the modulation of sleep/alertness, and mood [29]. The retinohypothalamic pathway, which mediates the retinal melanopsin stimulation into neuronal signals to the SCN, is known to become dysfunctional in age-related ocular and neurodegenerative diseases such as glaucoma and Parkinson’s disease due to mRGC loss [13]. The loss of function of these cells disrupts the circadian rhythm and could be a potential cause of sleep disruption. Chromatic pupillometry (using different wavelength light stimulus) has the potential to assess the functions of mRGCs in vivo, as mRGCs have the highest sensitivity to blue light at 480 nm [30, 31]. Although measuring the pupil response post-illumination is considered an accurate metric of mRGC function [32], the use of chromatic pupillometry in the elderly population with mixed neurodegenerative pathologies, including AD, needs to be further investigated.
Moreover, the retina has been used as a therapeutic target for modulating sleep. Hanford et al. review studies completing bright light treatment in AD or related dementia that light treatment could improve the duration of the sleep period, increase daytime wakefulness, and reduce evening agitation [33]. Similarly, in a sample of 29 institutionalized AD individuals aged 60–98 years, Dowling et al. showed that using one-hour of light intervention Monday through Friday for 10 weeks improved sleep efficiency, sleep duration and stabilized the most active hours outside of typical hours of sleep in individuals who had severe sleep disruptions despite the nonsignificant improvement compared to the controls [34]. In a sample of 189 dementia individuals living in assisted care facilities (120 with AD) aged 80–91 years, Riemersma-van der Lek et al. identified that bright light and melatonin were able reduce cognitive decline, reduce nocturnal restlessness, increase sleep duration and efficiency, and counteract negative impact of melatonin on mood and behavior [35]. Thus, retinal exposure to light may play a crucial role in the sleep-wake function and circadian rhythm function.
There are few studies examining melatonin in preclinical AD. However, in people with MCI, Naismith et al. showed that there was advanced timing of melatonin secretion compared to control subjects [26]. Subsequent studies have also shown advanced timing in people with subjective cognitive complaints [36]. A systematic review by Nous et al. analyzing the limited number of studies analyzing levels of melatonin in CSF, blood, saliva, and urine in AD concludes that there is a decrease in nighttime melatonin concentrations [37]. In postmortem AD, Liu et al. demonstrates there is a significant decrease in melatonin levels in the CSF, independent from a decrease seen with age [38]. Trials of melatonin also show promise in improving sleep, cognition, and mood [39–41]. Whether melatonin supplementation has any beneficial effects in at-risk individuals such as those with MCI, preclinical AD or subjective cognitive decline is unknown, but initial trials are underway in this regard (Schrire et al. [42], 2021). While animal studies suggest that melatonin may target Aβ deposition, human randomized controlled trials have yet to determine whether there are beneficial effects on Aβ, tau, oxidative stress, or other neurodegenerative markers.
SLEEP IN AD
The mechanisms behind sleep disruptions in AD are not yet fully understood. Variability in the sleep-wake cycle is common in individuals with AD [43]. Ju et al. reported in a preclinical AD sleep study that participants with worse sleep quality had higher levels of brain Aβ deposition [44]. Specifically, in 145 cognitively normal individuals, the authors measured sleep using actigraphy for two weeks and examined sleep quantity, efficiency (i.e., amount of time spent in bed asleep and total time asleep), and brain Aβ levels. One proposed mechanism for sleep variability is the loss of neuronal cells in the SCN, as Stopa et al. identified with postmortem immunostaining in individuals with AD compared to healthy controls [45]. In a mouse model, Kang et al. demonstrates the circadian fluctuation of Aβ in the extracellular fluid, increased levels during wakefulness and sleep deprivation, and orexin agonists reduce Aβ levels [46]. Roh et al. also identified in a mouse model of AD, Aβ plaques in the brain caused a deterioration in the levels of Aβ in the interstitial fluid and the sleep-wake cycle [47]. In a review study, Wu et al. reported that sleep deprivation could impair the clearance of brain Aβ and tau, increase oxidative stress in the brain and reduce melatonin levels, and that this association is bidirectional [48]. Variability in sleep disruptions in AD could be attributed to its role in influencing the expression levels of Aβ in the brain.
It is also unclear whether there is a critical period by which sleep disturbance may have a deleterious effect on brain structure, function, and Aβ accumulation. In a UK study, sleep disturbance at midlife was predictive of dementia 25 years later [49]. In another study using the European Prevention of Dementia dataset, there were differential relationships between various sleep disturbances and CSF-derived AD pathology at midlife, older age, and late-life [50]. Using PET to evaluate aquaporin-4, a brain Aβ clearance mechanism, in a sample of 222 cognitively normal participants aged 69–81 years, Rainey-Smith et al. identified variants and single-nucleotide polymorphisms that moderate the association of poorer overall sleep quality, latency and duration on brain Aβ burden [51]. In a sample of 184 cognitively healthy participants aged at least 60 years, Brown et al. evidenced sleep factors including longer sleep latency are associated with higher levels of brain Aβ [52]. Thus, in addition to evidence showing that neurodegeneration due to AD may contribute to sleep disturbance, there appears to be evidence that sleep disturbance precedes AD, as well as evidence that those with AD and neurodegeneration decline faster [53].
THE CROSSTALK BETWEEN SLEEP-RETINA-Aβ
A sleep-retina-Aβ link is plausible, as PLR of the eye and sleep regulation are both mediated by the retina’s population of mRGCs. Table 1 provides an overview of the studies examining the links between sleep, the retina, and AD. A possible mechanism for sleep-wake disruption in AD may be caused by the deposition of retinal Aβ surrounding the functional mRGCs, attenuating their role as regulators of circadian rhythms [17, 54]. La Morgia et al. performed OCT in AD (n = 21) and healthy age-matched controls (n = 74) to determine if there is an age-related loss of RNFL thickness [55]. Using actigraphy, the authors reported circadian rhythm dysfunction and reduced sleep efficiency in the AD group. Moreover, in postmortem analysis of horizontal retinal sections (AD n = 17, control n = 13) using antibodies targeting melanopsin, the authors reported axonal loss due to age, and a loss of mRGCs independent of age. Further analysis of the postmortem AD retinas revealed selective deposits of retinal Aβ surrounding the mRGCs in the superior region [55]. However, this is an early link as results have only been reported by one group, and independent investigations by other researchers are required to validate these findings.
Of interest, Oh et al. tested PLR as a function of the mRGCs in a preclinical cohort of AD to measure its suggested dysfunction [56]. The team did not find statistically significant differences between the AD subjects and control groups in terms of PLR function but noted the variability in sleep-wake function within the preclinical AD group [57].The authors suggested that there may be further subtypes of mRGCs to account for the preserved function of PLR and disruption in sleep-wake rhythms [57, 58]. Further investigation may help understand the link between mRGC function and AD symptom expression of disrupted sleep.
Moreover, there is emerging literature reporting a connection between obstructive sleep apnea (OSA), retinal health, and AD. Ferrandez et al. reported that peripheral vision function is reduced in individuals with OSA when compared to age-matched controls [59]. Brodie et al. and Schall et al. noted that individuals with OSA have a higher risk for retinal diseases such as diabetic retinopathy and age related macular degeneration (AMD) and the effectiveness of anti-vascular endothelial growth factor (VEGF) therapy treatment for AMD is reduced in individuals with OSA [60, 61]. Possible mechanisms associating OSA to retinal health includes high or transient changes in blood pressure due to OSA which increase sympathetic nerve stimulation and reduces blood flow to the eye, OSA-related hypoxia altering the chemical environment or high production of VEGF being expressed [60, 61]. Owen et al. reported that presence of tau within the hippocampus was independent of OSA severity, whereas Aβ deposition in the hippocampus was highly correlated to OSA severity [62]. The 2022 American Thoracic Society workshop; a multidisciplinary perspective on OSA, cognition, and dementia, strengthens the OSA and AD association as they note a heterogenous improvement in cognitive domains in studies of continuous positive airway pressure (CPAP) treatment [63]. Overall, the information and mechanisms explaining the bidirectional relationship of various types of sleep disruption in AD could provide surrogate biomarker evidence for early AD diagnosis in addition to its physiological link.
ROLE OF HORMONES
To further understand the mechanisms behind the crosstalk between Aβ, the retina, and sleep in AD, it is important to mention the role of certain circadian hormones linked to photoentrainment (synchronizing of circadian rhythm cycles to light and dark). Figure 3 illustrates the crosstalk between Aβ retina and sleep in AD and the involvement of hormones. Hormones such as melatonin, orexin, ghrelin, and leptin are associated with sleep regulation and their levels have been shown to be disrupted in AD. Furthermore, the resultant Aβ deposition in the retina can negatively affect retinal health. Melatonin is one of the direct modulators of sleep quality. Orexin is an arousal hormone and plays an important role in sleep regulation [64]. In sleep deprivation caused by narcolepsy, levels of orexin within the CSF have been found to be abnormal [64]. Similarly, in animal and cell model studies Liu et al. reported an increased expression of orexin receptors in the presence of Aβ, thereby, increasing the effectiveness of orexin causing arousal [65]. Ligouri et al. review orexin’s circadian regulation and the prevalence of its dysregulation due to AD, resulting in sleep impairment [66]. This review highlights a similar pattern of orexin dysregulation occurring in preclinical AD individuals. The circadian dysregulation causes disruption in the expression of orexin and melatonin levels and in turn cause alterations of the sleep cycle, secondarily affecting the clearance of Aβ [67]. Beyond the scope of this review, the role of transcriptomics and genetic information including the role of clock genes (period and cryptochrome genes) may further explain the complex mechanisms in the crosstalk between Aβ, sleep and retina. The proposed effect of AD on the retina’s health and circadian influence, in context of growing evidence of an ocular lymphatic and glymphatic system [68] may provide insight on AD and Aβ’s effect on sleep and the retinal presentation.
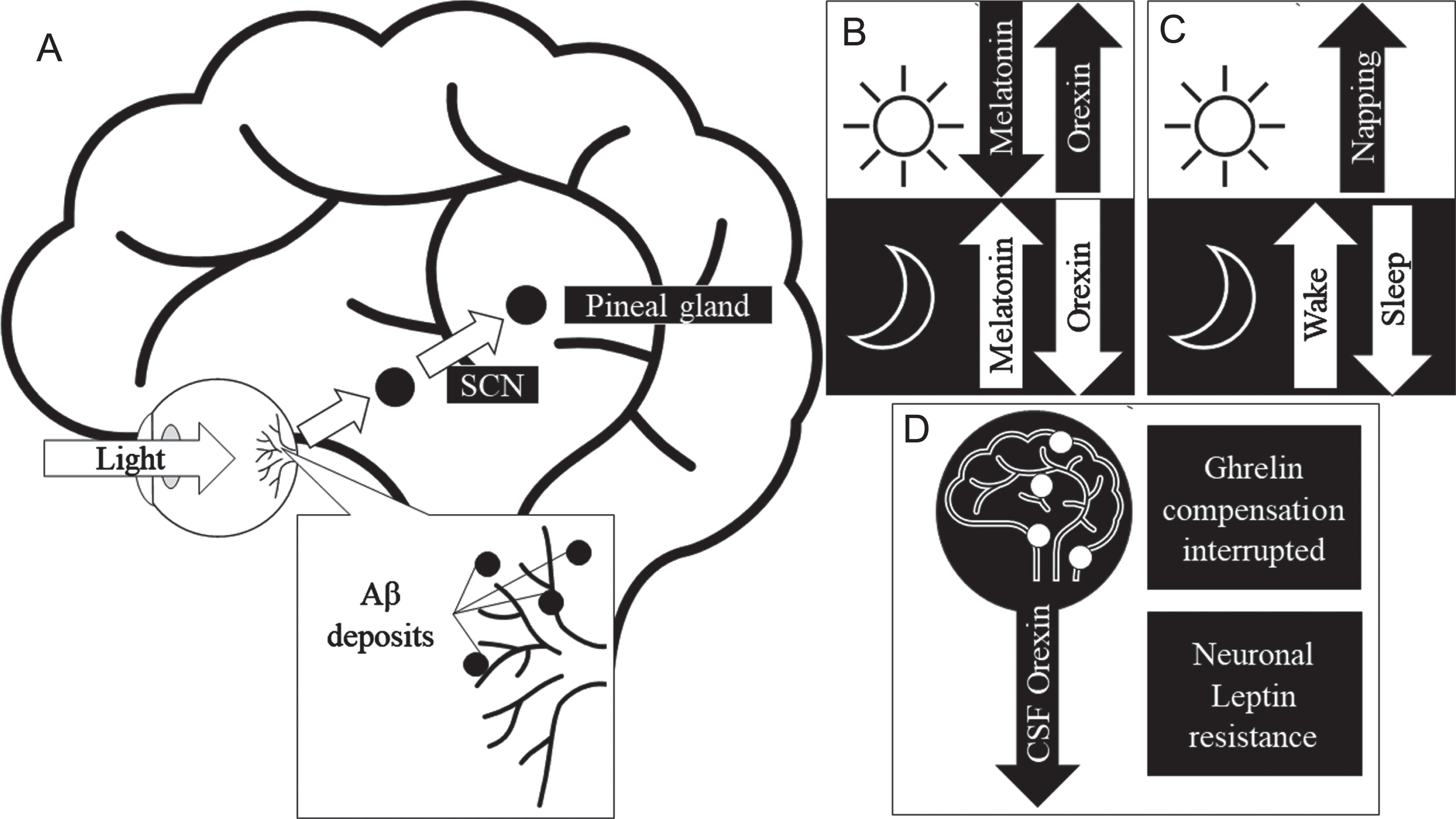
Proposed mechanism for Aβ in the retina to cause sleep-wake cycle and circadian hormone disruptions. A) Physiological pathway of circadian activity. B) Normal activity of sleep hormone expression in response to light saturation in day and night. C) Changes to sleep-wake patterns in response to AD, Retinal Aβ may disrupt the pathway observed in (A) in AD, thereby causing disruption in sleep quality. D) Circadian hormones effected in AD, where CSF orexin expression is decreased and the attenuation of ghrelin and leptin’s neuroprotective role. Aβ, amyloid-β; AD, Alzheimer’s disease; SCN, suprachiasmatic nucleus.
Other hormones of interest are ghrelin, and leptin which are known as hunger hormones [66]. These peptides are recognized as important players in metabolic dysfunction seen within AD individuals and have been successfully correlated to the risk of obesity; with excess body weight contributing to cognitive decline and developing AD [69, 70]. These hormones play additional peripheral roles within the hippocampus, promoting learning, and memory [71], whereby there is evidence leptin receptors are expressed within the hippocampus [72, 73]. Fewlass et al. have demonstrated that leptin treatment can modulate Aβ clearance [74] and therefore leptin deficient models are suggested to have poor spatial memory and downstream increases of brain Aβ [75]. Ghrelin supports the maintenance of neuronal synapse formation as Carlini et al. has demonstrated with dose injections of ghrelin at concentrations of 0.3, 1.5, and 3 nm within a WISTAR rat model and the step-down test, that memory retention increases are proportionate with the level of ghrelin [76]. Moreover, the Martins et al. group reported that both leptin and ghrelin improved hippocampal cell survival against Aβ oligomer toxicity [71]. Sleep deprivation can impact the expression of these hunger hormones as seen within normal circadian activity [77]. The outcomes of the study by Figueiro et al. demonstrate that the levels of both ghrelin and leptin in sleep restricted individuals are influenced by morning light, independent of other normal stimulants [78]. Retinal and sleep-wake interactions influencing hormone expression warrant further study; exploring within AD and preclinical AD cohorts may further understanding on the mechanisms which connect AD retinal pathology and sleep disruption.
CONCLUSION
Based on this review, it is suggested that the assessment of sleep, circadian rhythm and related hormones may have the potential to be used as surrogate markers along with changes identified using retinal imaging. The presence of sleep-wake disruption in AD, early, preclinical and MCI stages, are well evidenced in the literature suggesting that sleep-wake functions could be a viable early intervention target for risk factor modification. From the association between sleep and mRGCs, sleep and related hormone level disruptions may indicate retinal damage in AD might be used as a novel diagnostic marker. Current early diagnostic markers are invasive, expensive and cannot be used in a clinical setting, whereby imaging of the retina would allow us to non-invasively investigate brain health in AD. New methods such as hyperspectral imaging have clinical trials currently underway to address challenges associated with imaging the live human retina to identify signature AD biomarkers and thereby validate a simple eye test for population screening. There is a need for future studies using gold standards of measuring sleep neurophysiology to cross-sectionally and longitudinally evaluate the extent of the retinal influence on sleep-wake changes, cognitive decline and Aβ accumulation. Therefore, the associations, synergies and crosstalk between retinal health, sleep-wake functions and Aβ may further inform diagnosis and screening of preclinical and AD populations.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors acknowledge Macquarie University, the Australian Alzheimer’s Research Foundation, and the Lions Alzheimer’s Foundation for funding support.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.