
Abstract
Alzheimer’s disease (AD) is a lethal neurodegenerative disorder characterized by severe brain pathologies and progressive cognitive decline. While the exact cause of this disease remains unknown, emerging evidence suggests that dysregulation of neurotransmitters contributes to the development of AD pathology and symptoms. Serotonin, a critical neurotransmitter in the brain, plays a pivotal role in regulating various brain processes and is implicated in neurological and psychiatric disorders, including AD. Recent studies have shed light on the interplay between mitochondrial function and serotonin regulation in brain physiology. In AD, there is a deficiency of serotonin, along with impairments in mitochondrial function, particularly in serotoninergic neurons. Additionally, altered activity of mitochondrial enzymes, such as monoamine oxidase, may contribute to serotonin dysregulation in AD. Understanding the intricate relationship between mitochondria and serotonin provides valuable insights into the underlying mechanisms of AD and identifies potential therapeutic targets to restore serotonin homeostasis and alleviate AD symptoms. This review summarizes the recent advancements in unraveling the connection between brain mitochondria and serotonin, emphasizing their significance in AD pathogenesis and underscoring the importance of further research in this area. Elucidating the role of mitochondria in serotonin dysfunction will promote the development of therapeutic strategies for the treatment and prevention of this neurodegenerative disorder.
INTRODUCTION
Serotonin, also known as 5-hydroxytryptamine, is a member of the monoamine neurotransmitter family [1, 2]. In the central nervous system, serotonin is predominantly synthesized and released by the dorsal raphe nucleus, where serotonin-producing neurons reside [1, 2]. As a critical component of the diffuse nervous system, serotonin signaling contributes to the modulation of multiple key brain processes, including mood and emotion, cognition and memory, appetite and satiety, the sleep-wake cycle, and social interactions [3–10]. Accordingly, serotonin plays a pivotal role in brain health, with its dysregulation has been linked to a plethora of neurological and psychiatric disorders including Alzheimer’s disease (AD) [11–13]. While serotonin itself is not the underlying cause of AD, the contribution of serotonin signaling deregulation to the progression of symptoms reveals unique therapeutic opportunities; these serotonin signaling-targeting therapies having demonstrated substantial therapeutic benefits [14–16]. Therefore, elucidating the regulatory mechanisms of serotonin has a positive impact on our understanding of both neurobiology and neurodegeneration.
Mitochondria are vital organelles that support neuronal functions through their roles in producing energy, regulating calcium homeostasis, and maintaining oxidation-reduction (redox) balance [17, 18]. Mitochondrial dysfunction has been consistently identified as a key factor underlying synaptic injury and neuronal stress in AD, which has been extensively reviewed elsewhere [19–22]. Recently, not only has the impact of serotonin on mitochondrial fitness emerged, but mounting evidence also suggests the importance of mitochondrial function in the regulation of serotonin homeostasis [11, 23]. Reflecting the growing interest in the role of serotonin in neurological decline and wellbeing, this review aims to summarize recent research progress in the intertwined relationship between brain mitochondria and serotonin. Additionally, we discuss the association between mitochondrial dysfunction and serotonin dysregulation in AD and its therapeutic potential for the treatment of this devastating neurological disorder.
MITOCHONDRIA AND SEROTONIN REGULATION IN NEUROBIOLOGY
Mitochondrial function and neuronal serotonin transmission and recycling
Serotonin is synthesized through a two-step reaction. Firstly, tryptophan hydroxylation converts tryptophan into 5-hydroxytryptophan (5-HTP) by tryptophan hydroxylase isoform 2. This is followed by aromatic L-amino acid decarboxylase-mediated 5-HTP decarboxylation [24, 25]. In the presynapse, the synthesized serotonin is then immediately packaged into synaptic vesicles by the vesicular monoamine transporter (VMAT), especially VMAT2, and subsequently released into the synaptic cleft, where it interacts with serotonin receptors in the postsynaptic membrane [26]. A significant portion of the released serotonin is recaptured by presynapses via the serotonin transporter (SERT) [27, 28] and under further degradation by mitochondrial enzymes including monoamine oxidase isoform A (MAO-A) [29], thus completing the cycle of serotonin metabolism. Although the evidence supporting the direct involvement of mitochondria in serotonin synthesis is currently limited, the role of mitochondria in the regulation of serotonin transmission and breakdown is becoming increasingly evident.
VMAT2 is the major protein responsible for the loading of serotonin into synaptic vesicles for storage and subsequent release [26]. Previous studies have determined that the function of VMAT2 is fueled by the ATP-consuming V-type H+-ATPase [30] and regulated by phosphorylation modification [31, 32], both of which are ATP-dependent processes. Additionally, synaptic-vesicular transport requires high energy expenditure [33]. Given that mitochondria are the primary providers of ATP in the presynapse [34], it is plausible that mitochondrial ATP production is crucial for serotonin storage and transport. Indeed, in our recent study, we have observed impaired serotonin release in mouse hippocampal slices subjected to stress induced by carbonyl cyanide-p-trifluoromethoxy phenylhydrazone, a mitochondrial uncoupling agent [11]. These findings suggest that mitochondrial function plays an integral role in serotonin release and support the notion of mitochondrial involvement in serotonin neurotransmission. Moreover, the activity of SERT relies on the maintenance of a cross-membrane Na+/K+ gradient, which is created by the energy-consuming Na+/K+ ATPase pumps [35]. As a result, impaired mitochondrial bioenergetics could also potentially compromise serotonin recycling and reuptake processes.
It is worth noting that mitochondria serve as a stabilizer of Ca2+ homeostasis in neurons [36, 37]. The VMAT2-mediated filling and release of serotonin within synaptic vesicles involve Ca2+ signaling, including the participation of Ca2+-dependent activator proteins of secretion 1 and 2 [38]. Furthermore, the exocytosis of synaptic vesicles is modulated by Ca2+ signaling, which can be influenced by mitochondrial sequestration of Ca2+ [39]. In this regard, mitochondrial regulation of Ca2+ may also contribute to serotonin transmission, and impaired mitochondrial Ca2+ retention capacity can impede serotonin regulation. The direct link between mitochondrial Ca2+ handling capacity and serotonin release is determined in mice with heterogeneous loss of mitochondrial adenine nucleotide translocase 1 (ANT1) [40]. Serotonin-producing cells in the dorsal raphe nucleus demonstrate susceptibility to ANT1 deficiency-induced mitochondrial dysfunction, which coincides with dorsal raphe neuronal hyper-depolarization and resultant increases in serotonin turnover [40]. Due to the involvement of ANTs including ANT1 in the regulation of mitochondrial Ca2+ through mitochondrial permeability transition [41], it is proposed that the influence of this mitochondrial protein deficit on serotonin regulation is, at least in part, associated with Ca2+-related perturbations [40]. These findings therefore highlight the need for further comprehensive investigations into the role of mitochondrial Ca2+ handling in serotonin signaling.
Mitochondrial enzymes and serotonin degradation
MAO-A, an isoform of MAO, is an outer mitochondrial membrane-bound enzyme that converts serotonin to its metabolite, 5-hydroxy-3-indolacetaldehyde (5-HIAL) [42]. This toxic biogenic aldehyde is further processed into the less toxic 5-hydroxy-3-indolacetic acid by a mitochondrial matrix protein, aldehyde dehydrogenase 2 (ALDH2), for excretion [28, 43]. Previous studies have consistently reported that loss of function of MAO-A reduces serotonin breakdown in the brain, leading to behavioral abnormalities in humans and rodents [44–49]. Furthermore, suppressed ALDH2 activity disrupts serotonin metabolism, resulting in the accumulation of the toxic serotonin metabolite 5-HIAL and leading to pathological consequences [28, 51]. The significance of these mitochondrial enzymes in serotonin metabolism thus underscores the key role of mitochondria in maintaining neuronal serotonin homeostasis.
SEROTONIN AND MITOCHONDRIAL REGULATION IN NEUROBIOLOGY
Despite the role of mitochondria in regulating serotonin transmission and metabolism, the impact of serotonin on neuronal mitochondrial fitness has been accentuated in recent years [23, 53]. A recent finding demonstrated that serotonin induces mitochondrial biogenesis in mouse cortical neurons [23]. Such a mitochondrial biogenesis-promoting effect of serotonin is achieved through the stimulation of serotonin receptor 2A (5-HT2A), the receptor’s downstream signaling involving sirtuin 1, and the peroxisome proliferator-activated receptor gamma coactivator 1 alpha [23]. Interestingly, serotonin signaling-induced mitochondrial biogenesis and subsequent alterations in cellular metabolism, favoring increased oxidative phosphorylation, have also been reported in breast cancer [54]. This suggests that the role of serotonin in regulating mitochondria is not exclusive to neural cells. Additionally, the influence of the SERT on mitochondria has been reported [55]. A previous study showed a negative correlation between SERT expression and mitochondrial copy number in the brains of male rats, while reduced SERT expression led to a decrease in mitochondrial copy number in females [55]. Regardless of the unclear mechanisms underlying the impact of SERT on mitochondria and the sex-related dimorphic regulation, these findings implicate the involvement of serotonin signaling in mitochondrial biology. Moreover, Reddy and the colleagues in a recent study reported that citalopram, a selective inhibitor of SERT, promotes mitochondrial fusion, potentiates mitochondrial generation, and enhances mitochondrial turnover through mitophagy in a mouse hippocampal cell line [56]. Although it cannot be excluded the non-SERT-related off-target effects of citalopram on mitochondria, these results echo the impact of SERT expression modulation on mitochondrial regulation [55], further supporting the potential association of serotonin signaling with mitochondrial fitness even in non-serotonin-producing neural cells.
MITOCHONDRIA AND SEROTONIN IN ALZHEIMER’S DISEASE
Serotonin deficiency in AD
AD is a chronic neurodegenerative disorder characterized by pathological features, including amyloid-β (Aβ) aggregation, abnormal tau phosphorylation, synaptic and neuronal degeneration. In addition to the defining manifestation of progressive memory loss, patients with AD frequently demonstrate psychiatric symptoms, including mood and emotional fluctuations, social behavioral changes, as well as sleep disturbances [57–64]. Both depression and anxiety have been repeatedly identified in patients with AD or even in the prodromal stage of AD [65–70]. Previous studies reported depressive and anxiety symptoms in a significant portion of patients in different stages of AD with mild to severe cognitive deficits [71–73]. Furthermore, meta-analytic studies have identified mood disorders including depression and anxiety as strong risk factors for the development of AD [74–79]. Because of the well-established central role of serotonin dysregulation in mood disturbances including depression and anxiety problems [80–83], the strong negative association of depression and anxiety with cognitive performance in AD patients determined in previous clinical studies [68, 70] justifies further investigations of serotonin dysregulation in this neurodegenerative disorder. Although AD is not a typical neurotransmitter disorder, serotonin deficiency in the brains of AD patients has long been observed to be associated with mood disturbances and cognitive impairment [84, 85]. The loss of serotoninergic neurons in the raphe nucleus was first reported four decades ago and has been consistently observed to coincide with reduced serotonin and its metabolites since then [85–93]. The lesions in the raphe nucleus in patients in the early stage of AD further implicate an association of serotonin dysregulation with the development of this neurodegenerative disorder [94, 95]. Consistent with the findings in patients [85, 96–98], a reduction in serotonin-producing neurons in the raphe nucleus and a decrease in serotonergic fiber density in multiple brain regions including the neocortex and hippocampus has been determined in several mouse models of AD-like brain amyloidosis [11, 99]. These findings suggest detrimental impacts of Aβ on the fitness of serotonergic neurons. In addition to Aβ deposition, abnormal tau aggregation constitutes another AD-related pathological characteristic [100]. Previous studies have also determined neurofibrillary tangles composed of hyperphosphorylated tau in the dorsal raphe nucleus in AD patients [101–104] and proposed an association of tauopathy with AD-related serotoninergic neuron degeneration and serotonin deprivation [104, 105]. The direct link between tau abnormalities and serotonin dysregulation was determined in further animal experiments. Marcinkiewcz’s group showed that overexpression of human tau in mice induces degenerative changes of serotonin-producing neurons [106]. Accordingly, tau depletion prevents chronic stress-mediated mouse brain serotonin loss [107]. Moreover, manipulating serotonin signaling reciprocally promotes the development of tau aggregation [108, 109]. These findings in together highlight the interactions between serotoninergic system and tau pathology. However, a previous study reported a paradoxical increase in hippocampal serotoninergic fibers in 3xTg mice, another familial AD mouse model with brain amyloidosis and tauopathy [110]. This discrepancy between 3xTg mice and other mouse models with brain amyloidosis and/or tauopathy may reflect differences in the ability of different mouse models to recapitulate AD pathologies, raising interesting questions regarding mouse model selection for studying AD-related disruption of the serotoninergic system. Of note, despite the deleterious impact of Aβ and tau on serotoninergic neurons, we cannot fully refute the possibility that tau and Aβ or amyloid-β protein precursor may have unknown interference with each other’s effect on serotoninergic neurons, impeding the degeneration of these neurons in 3xTg mice. This needs further investigation. In addition to perturbations in serotonin and serotoninergic neurons, lowered levels of the SERT have been identified in multiple brain regions in patients at various stages of AD [85, 91]. Of note, some serotonin-enhancing agents such as citalopram also display a Aβ-lowering capability [56, 112], possibly through the activation of serotonin receptors including 5-HT2 R and 5-HT4 R [113, 114]. Moreover, serotonin receptor modulators also demonstrate therapeutic effects against tau pathology [108]. In addition to the therapeutic benefits observed with selective serotonin reuptake inhibitors (SSRIs) and serotonin receptor modulators in AD patients and animal models [14–16, 115–117], clinical studies have also found protective effects of SSRIs against cognitive decline and cortical atrophy in older adults with concurrent mild cognitive impairment and depression [15]. Therefore, these findings strongly demonstrate serotonin dysregulation in the etiopathogenesis of this neurodegenerative disorder.
Mitochondria dysfunction and serotonin deficiency in AD-related conditions
Although mitochondria dysfunction has been well-documented in AD paradigms [19, 118–120], previous studies on mitochondrial dysfunction in AD overwhelmingly focused on mitochondria in pyramidal neurons in the neocortex and hippocampus. It should be noted that, despite the vulnerability of the limbic system to neuronal stress, degenerative changes and AD-associated pathologies in the brain stem, including the serotonin-producing raphe nucleus, are also prominent in patients with AD [93, 97]. To this end, although the investigation of mitochondrial function in serotoninergic neurons in AD-related conditions has been seldom explored, the mitochondrial sensitivity to AD-associated pathologies and the reported deleterious impacts of mitochondrial dysfunction on raphe neuron functions [40] thus warrant a hypothesis of a role of mitochondrial dysfunction in serotoninergic neuronal stress in this neurodegenerative disorder.
Hippocampal lesions are a key pathological feature underlying the symptoms of AD [121–123]. The hippocampus, a vital brain region involved in memory storage, information processing, and mood modulation [124–127], heavily relies on serotonin for its optimal functioning [128–130], aligning with the findings that serotoninergic neurons innervate the neurons in the hippocampus, and all types of serotonin receptors are present in this region [131, 132]. SSRIs, a class of medications that enhance serotonin levels, have shown therapeutic effect in alleviating AD symptoms [14–16, 115–117], further emphasizing the impact of serotonin dysregulation on hippocampal-related cognitive and mood disturbances associated with the disease. Our recent study has provided evidence of concurrent impairments in hippocampal serotonin release and mitochondrial morphological control within hippocampal serotoninergic fibers in 5xFAD mice, a mouse model of familial AD-like brain amyloidosis [11]. Serotoninergic neurons in Aβ-rich environments exhibited reduced mitochondrial bioenergetics and increased mitochondrial fragmentation [11]. Additionally, the administration of a mitochondria-uncoupling agent resulted in the suppression of serotonin release in hippocampal slices [11]. Considering the significance of mitochondrial health in serotonin transmission, these findings offer important insights into the contribution of mitochondrial dysfunction to serotonin dysregulation in this neurodegenerative disease, warranting further investigation into the role of mitochondrial dysfunction in raphe nucleus neurodegeneration within AD-relevant pathological settings. Moreover, given the role of serotonin in promoting neuronal mitochondrial biogenesis, there may exist a vicious cycle of mitochondrial dysfunction and serotonin failure that reinforces each other, leading to damage in both serotoninergic and non-serotonergic neurons in AD brains.
Other than the direct influence of mitochondrial dysfunction on serotonin transmission, changes in MAO, a mitochondrial enzyme responsible for serotonin metabolism, have been implicated in AD-relevant settings. In addition, altered MAO activity has a correlation with AD neuropsychiatric symptoms and pathological changes, including amyloid deposition and neurofibrillary tangles [133–137]. So far, there is no evidence of the impact of MAO alterations on serotonin metabolism in AD brains. However, the administration of MAO inhibitors does exhibit clinical benefits to some extent in mitigating AD symptoms, including psychiatric symptoms [138, 139]. Although serotonin is not the sole substrate of MAO [29], the therapeutic effect of MAO inhibitors in alleviating psychiatric symptoms arguably implies an influence of MAO changes on serotonin signaling in AD. Nevertheless, declining serotonin metabolites in AD patients may not solely arise from lowered serotonin synthesis, but also from enhanced serotonin degradation by MAO. In this context, we cannot refute the possibility that MAO, especially MAO-A, is also hyperactive in serotoninergic neurons, leading to pathological consequences such as serotonin degradation and subsequent disturbances in serotonin signaling in AD-related conditions. However, it should be noted that a previous study reported a simultaneous decline in both serotonin and MAO-B in platelets from very late-stage AD patients. So far, there are mixed results regarding MAO-B expression in neural cells. MAO-B expression has been previously identified in monoaminergic including serotoninergic neurons [140–143]. In contrast, other reports including a recent neuroimaging study showed that MAO-B is not expressed by neurons [144, 145]. Regardless of the yet-unsettled question of MAO-B expression in neurons, there is no evidence of a role of MAO-B in neuronal serotonin metabolism; and MAO-A is the only genetically determined MAO family member to be responsible for serotonin degradation to date [146]. In this regard, the concurrence of platelet serotonin and MAO-B reduction may have limited capacity to indicate serotonin metabolism in AD platelets but is rather a reflection of systemic degenerative changes in the end stage of this neurodegenerative disorder. Given the strong relevance of MAO-A to serotonin regulation, the findings of the negative impact of MAO-A activity on mitochondrial bioenergetics in cortical neurons [147] further strengthened the hypothesis that MAO-A hyperactivity may exacerbate mitochondrial dysfunction, resulting in a downward spiral of worsened serotonin dysregulation in AD.
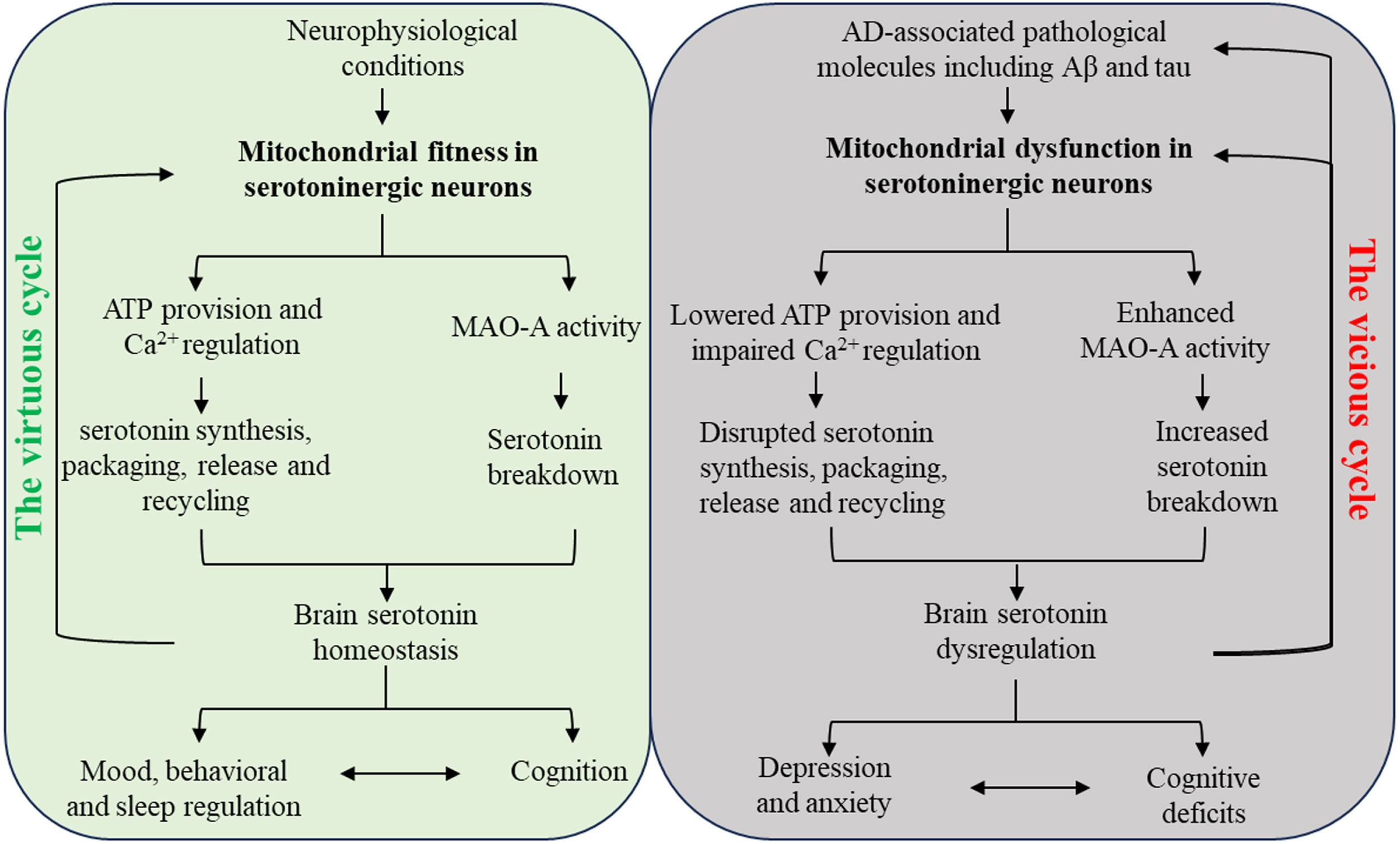
CONCLUSIVE REMARKS AND FUTURE PERSPECTIVES
In conclusion, the interplay between mitochondria and serotonin signifies a complex relationship between these two entities with substantial implications for neurobiology. In the context of AD, this delicate balance between serotonin and mitochondrial function can be disrupted, giving rise to a vicious cycle of mitochondrial dysfunction and serotonin failure that mutually reinforce each other, resulting in pathological consequences in this neurodegenerative disorder (Fig. 1). There are several outstanding questions that remain unresolved. It is unclear whether mitochondrial dysfunction or serotonin dysregulation in raphe neurons occurs first during the development of this age-related disorder. Of note, the scientific community has yet to reach a consensus on whether raphe neurons are sensitive to the aging process has not yet reached a consensus [148, 149]. However, it is commonly accepted that aging renders neuronal mitochondria vulnerable to functional deficits [150–154]. In this regard, it can be hypothesized that age-related mitochondrial dysfunction is an initiating factor for the damage of raphe neurons, leading to serotonin failure in the elderly at risk of AD. However, we cannot fully exclude the possibility that mitochondrial dysfunction or serotonin dysregulation in raphe neurons develops independently and exacerbates each other with AD progression, given the yet-elusive complicated regulation of serotonin metabolism and early demonstration of raphe nucleus degeneration in early AD [94, 95]. Another question that merits consideration is the influence of disrupted serotonin and mitochondrial interactions on the development of neuroinflammation in AD brains. Growing evidence suggests that both serotonin dysregulation and mitochondrial dysfunction contribute to inflammatory neuronal damage [155–160]. In this context, the investigation of serotonin and mitochondrial dysfunction should not be limited to neurons but should also be extended to glial cell pathology in AD. Furthermore, mitochondria in neurons within the diffuse modulatory nervous system have long been neglected in the study of mitochondrial dysfunction in AD. It is important to recognize that dysfunction in serotoninergic, dopaminergic, and cholinergic neurons within the diffuse modulatory nervous system also play a significant role in AD-associated pathology and contribute to the development of the disease [84, 161–163]. Therefore, there is an urgent need for neuron type-specific investigations to comprehensively understand the mitochondrial pathways involved in this devastating neurological disorder. Lastly, it is increasingly recognized that serotonin-modulating therapies, especially SSRIs, demonstrate racial and ethnic disparities in their efficacy [164, 165]. Moreover, SSRIs and serotonin–norepinephrine reuptake inhibitors (SNRIs) may risk the elderly to increase adverse cardiovascular events [166, 167]. Given these critical concerns about SSRIs and SNRIs, targeting mitochondrial dysfunction may hold promise to be a better strategy for the restoration of brain serotonin homeostasis for the amelioration of AD cognitive and mood symptoms.
In summary, understanding the complex relationship between mitochondria and serotonin is essential for unraveling the mechanisms behind serotonin dysregulation in AD. Further research is needed to explore these mechanisms and their potential as therapeutic targets to restore serotonin homeostasis and alleviate AD symptoms. The findings presented in this review underscore the significance of developing novel therapeutic strategies that target both mitochondrial dysfunction and serotonin dysregulation for the prevention and treatment of AD.
Footnotes
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This work was supported by research funding from Brightfocus Foundation research grant A2022036 S to LG, KU career development grant 2302009 to LG, NIH P30 AG072973 to the University of Kansas Alzheimer’s Disease Research Center’s Research Education Component, and REC fellowship to JT.
CONFLICT OF INTEREST
The authors have no competing interests to disclose.