
Abstract
Sirtuin-1 (Sirt1), encoded by the SIRT1 gene, is a conserved Nicotinamide adenine dinucleotide (NAD+) dependent deacetylase enzyme, considered as the master regulator of metabolism in humans. Sirt1 contributes to a wide range of biological pathways via several mechanisms influenced by lifestyle, such as diet and exercise. The importance of a healthy lifestyle is of relevance to highly prevalent modern chronic diseases, such as Alzheimer’s disease (AD). There is growing evidence at multiple levels for a role of Sirt1/SIRT1 in AD pathological mechanisms. As such, this review will explore the relevance of Sirt1 to AD pathological mechanisms, by describing the involvement of Sirt1/SIRT1 in the development of AD pathological hallmarks, through its impact on the metabolism of amyloid-β and degradation of phosphorylated tau. We then explore the involvement of Sirt1/SIRT1 across different AD-relevant biological processes, including cholesterol metabolism, inflammation, circadian rhythm, and gut microbiome, before discussing the interplay between Sirt1 and AD-related lifestyle factors, such as diet, physical activity, and smoking, as well as depression, a common comorbidity. Genome-wide association studies have explored potential associations between SIRT1 and AD, as well as AD risk factors and co-morbidities. We summarize this evidence at the genetic level to highlight links between SIRT1 and AD, particularly associations with AD-related risk factors, such as heart disease. Finally, we review the current literature of potential interactions between SIRT1 genetic variants and lifestyle factors and how this evidence supports the need for further research to determine the relevance of these interactions with respect to AD and dementia.
INTRODUCTION
Silent information regulator 1 (Sirt1) protein, a member of the sirtuin family of class III histone deacetylases, has been found to contribute to a wide range of biological regulatory pathways via its role in epigenetic regulation (e.g., acetylation and methylation) of genes related to cell growth, inflammation, apoptosis, mitochondrial biogenesis, neurobiological processes, cell senescence, and longevity [1]. Additionally, an age-related reduction in serum Sirt1 protein in multiple tissues seems to be involved in age-related disorders [2], including Alzheimer’s disease (AD).
AD, the most common neurodegenerative cause of dementia, is clinically defined by loss of short-term memory, impaired judgement and problem solving, and changes in behavior and mood [3]. It is characterized by the accumulation of neurofibrillary tangles (NFTs), composed of hyperphosphorylated tau protein, and extracellular amyloid-β (Aβ) plaques, in the brain. Moreover, significant neuronal loss in the hippocampus and entorhinal cortex with substantial gliosis has been demonstrated. The onset of these pathological features of AD can occur up to two decades before symptoms appear [4]. Early-stage detection of AD is difficult and there is a lack of effective treatment, meaning individuals living with the condition often require a long period of nursing care, resulting in substantial economic burden [5]. As such, improved early detection and an increased understanding of risk factors at preclinical stages is critical for effective prevention and treatment.
There is a large body of literature describing the role of genetics and environmental/lifestyle factors as modulators of the pathological mechanisms of AD and dementia. Recently, much research has focused on the potential interaction between these factors. Environmental factors, and in particular lifestyle factors, are often modifiable, for example, through changes to diet, physical activity, and other behaviors. Therefore, understanding how their effect on AD interacts with an individual’s genetic background has the potential to enable individualized lifestyle interventions, which may provide more effective prevention or treatment [6–8]. In the current review (schematic summary Fig. 1), we will first briefly introduce the Sirtuins, particularly Sirt1 and preliminary evidence to support its relevance to AD. We then discuss the current evidence linking Sirt1 with AD in more detail, including AD-linked pathological mechanisms and biological processes associated with AD. In the case of biological processes, this association with AD is either through genetic studies or through relevance to the disease. Next, we will review what links several lifestyle factors, of relevance to AD, to Sirt1/SIRT1, before discussing the current literature linking genetic variation within the SIRT1 gene with AD and different AD-related risk factors and co-morbidities. Finally, we summarize evidence for the potential interaction between SIRT1 genetic variants and lifestyle factors, with reference to chronic diseases considered as potential risk factors for dementia and AD.
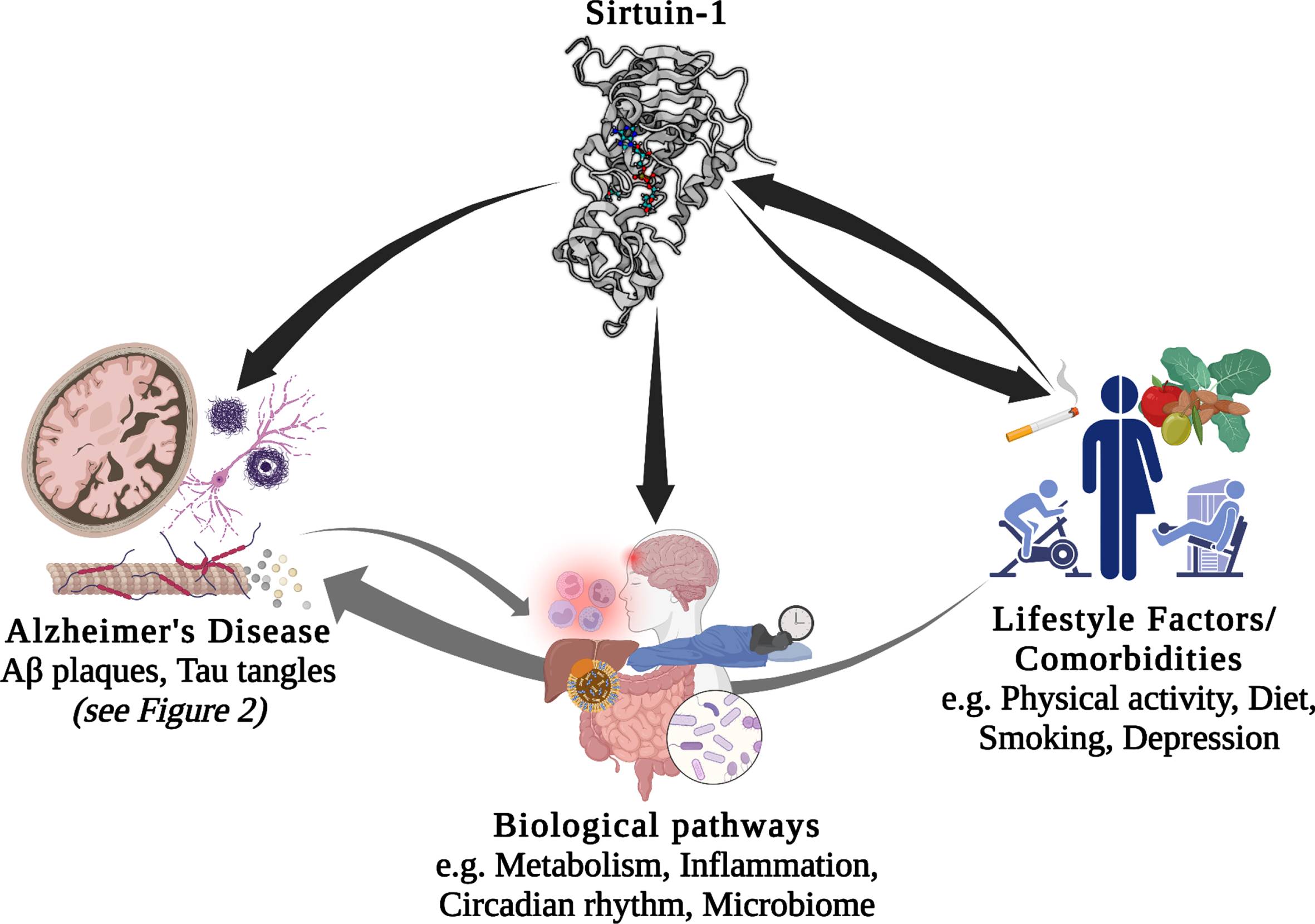
Potential interactions between Sirt1/SIRT1, lifestyle factors, comorbidities, biological pathways, and Alzheimer’s disease. Aβ, amyloid-β.
THE SIRTUINS AND SIRT1
Proteins in the sirtuin family are promising targets for improving aging phenotypes and ameliorating age-related diseases [9]. Seven members of the Sirtuin family of enzymes, are nicotinamide adenine dinucleotide (NAD)+-dependent class III histone deacetylases, and contribute to post-translational modifications (PTMs) of both nucleic and cytoplasmic proteins. Sirtuins remove acetyl groups from lysines through NAD; therefore, they contribute to a wide range of physiological processes, including regulating gene expression, metabolism, and senescence [1]. Sirt 6 and 7 are located in the nucleus, Sirt 2 in the cytoplasm, and Sirt 3–5 are in mitochondria. Sirt1 is found in both the nucleus and cytoplasm [10].
Among sirtuins, Sirt1 is the most studied and is expressed in adult brain, where it is predominantly found in neuronal nuclei [11]. Sirt1 can catalyze deacetylation of site-specific lysine residues of both histone and non-histone proteins, providing a potential connection between epigenetics, metabolism, and health status. Specifically, the NAD+/NADH ratio is a potential link between metabolism and epigenetic regulation of gene expression by contributing to histone acetylation [12]. Histone acetylation is an important epigenetic mechanism that alters chromatin structure and modulates gene expression by opening or closing the chromatin, playing a significant role in cell cycle and cell differentiation [13]. In culture media, SIRT1 overexpression improves dendritic spine density, neurite outgrowth and survival, and influences plasticity via brain-derived neurotrophic factor (BDNF) [14]. Neuroprotectivity of Sirt1 seems to be mediated by acetylation of neuronal p53, which suppresses induction of apoptosis [14]. Further, it has been suggested that Sirt1 is deployed to DNA damage sites to repair DNA, decreasing its activity in other loci of the genome and leading to “transcriptional noise”, which is a hallmark of cellular senescence [15]. Moreover, the activity of Sirt1 enzymes decreases with age [2], making it a good candidate biomarker for the study of age-related diseases, including AD [16].
SIRT1 AND ALZHEIMER’S DISEASE
AD pathological mechanisms
The contribution of Sirt1 to AD pathological mechanisms has been proposed in several studies. Sirt1 plays important roles in both Aβ and tau metabolism through various biological mechanisms (Fig. 2). It has been suggested to modulate the key enzymes/enzyme complexes integral to the metabolism of the amyloid-β protein precursor (AβPP), namely beta-secretase 1 (BACE1) and α-secretase (ADAM10, A Disintegrin and metalloproteinase domain-containing protein 10) [9], and thus may play an important role in regulating Aβ production. Studies of AD cases showed that in brain regions with Aβ accumulation, BACE1, which is the rate-limiting enzyme in Aβ metabolism is overexpressed [17]. Expression of BACE1 has been suggested to be regulated by peroxisome proliferator-activated receptor gamma (PPARγ), and its coactivator (PGC-1α), which when deacetylated by Sirt1, reduces BACE1 expression [17]. Moreover, ADAM10 cleaves AβPP to produce soluble AβPP (sAβPP), a neuroprotective fragment. The upregulation of ADAM10, through the deacetylation of retinoic acid receptor beta (RARβ) by Sirt1, could potentially protect the brain from Aβ accumulation through the potentiating the non-amyloidogenic cleavage pathway of AβPP and, in turn through increasing sAβPP, provides a neuroprotective mechanism against AD [18]. This is further supported by a photobiomodulation therapy study that showed that the activation of Sirt1 was responsible for the induced effects of reduced Aβ production, with knocking down of SIRT1 resulting in an antagonistic effect [19]. Taken together, there is growing evidence to support the key-role of Sirt1 in influencing the function of ADAM10 in the context of AD.
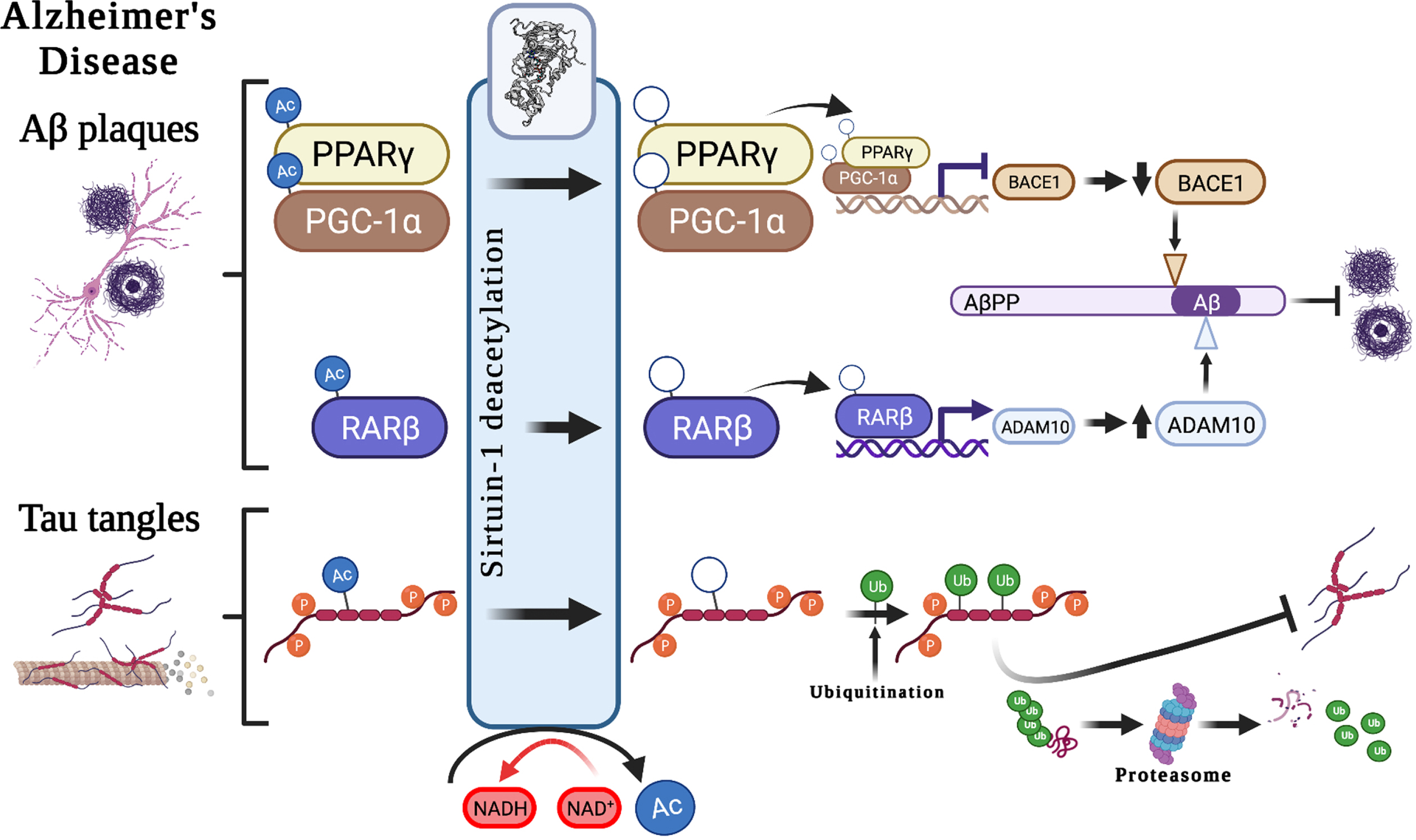
Potential associations between Sirt1 and Alzheimer’s disease hallmark pathological features. Suggested roles that Sirt1-mediated deacetylation plays in the development of the pathological hallmarks of Alzheimer’s disease, including the development of Aβ plaques and neurofibrillary tangles, through the modulation of AβPP cleavage pathways and tau degradation. Aβ, amyloid-β; AβPP, amyloid-β protein precursor; Ac, acetyl-; ADAM10, A Disintegrin and metalloproteinase domain-containing protein 10/α-secretase; BACE1, beta-secretase 1; NAD+, Nicotinamide adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide; PPARγ, Peroxisome proliferator-activated receptor-gamma; PGC-1α, peroxisome proliferator-activated receptor-gamma coactivator 1α; p, phospho-; RARβ, Retinoic acid receptor beta; Ub, ubiquitin.
Sirt1 also has autophagy-enhancing effects, thereby contributing to Aβ degradation [20]. In mammals, polyubiquitination can occur through autophagy–lysosomal pathways at lysine residues, interfering with acetylation. It is through this mechanism that decreased Sirt1 levels may result in hyperacetylation of substrates, such as phosphorylated tau [21]. Acetylated tau (ac-tau) has been reported to be increased in individuals with early and moderate Braak stages of tauopathy (a postmortem method for staging of AD progression based on anatomical localization of pathological features [22]), at earlier stages than the formation of NFTs. Moreover, Sirt1 has been shown to remove acetyl groups from tau protein, and an in vivo study showed Sirt1 deficiency increased ac-tau levels. As such, the inhibition of Sirt1, or the decline in Sirt1 enzyme activity observed with age [2], may result in a reduction in the deacetylation of tau, which in turn might block tau polyubiquitination and tau proteasomal degradation and lead to p-tau accumulation and, finally, the formation of NFTs [21].
Evidence for the importance of Sirt1 in memory and learning, the cognitive processes that are impaired in AD, is also emerging. Spatial learning and both short-term and long-term memory were disrupted n male mice, aged 5-6 months, with a brain-specific SIRT1 gene knockout [23, 24]. Although the brain for these mice had normal morphology and dendritic spine structure they showed a decrease in dendritic branching, branch length, and complexity of neuronal dendritic arbors. Also, there was downregulation of hippocampal genes contributing to synaptic function, lipid metabolism, and myelination. These findings provide evidence to support the important role of Sirt1 in neuronal development and plasticity. Moreover, epigenetic mechanisms such as DNA methylation contribute to memory formation and maintenance suggesting, Sirt1 may make a significant contribution through its role as an epigenetic “coordinator” [25].
Genetically linked AD biological pathways
Genome-wide association studies (GWAS) and analyses of gene expression data have identified multiple pathways associated with AD. Four such pathways include those responsible for cholesterol metabolism, immune response, regulation of endocytosis, and protein ubiquitination. Sirt1 is also known to modulate these biological pathways, particularly through deacetylation of many non-histone proteins, such as transcriptional nuclear receptors, that are involved in metabolism, inflammation, and apoptosis, and potentially age-related diseases such as AD [26]. The transcriptional regulatory proteins with the best evidence for modulation by Sirt 1 are PGC-1α, NF-κB (Nuclear factor kappa-light-chain-enhancer of activated B cells), p53 (tumor protein P53), FOXOs (Forkhead box O transcription factors), CRTC2 (CREB (cAMP response element-binding protein) Regulated Transcription Coactivator 2, LXRs (liver X receptors), FXR (farnesoid X receptor), and SREBPs (Sterol regulatory-element binding proteins) [27]. Here we review experimental evidence for Sirt1/SIRT1 activities in two key pathways regulating metabolism (specifically cholesterol metabolism) and inflammation, which we hypothesize may link Sirt1 to AD.
Sirt1 and metabolism
The deacetylation activity of Sirt1 links the enzyme to protein substrates, such as NAD [28, 29], an important coenzyme in all living cells. Through electron transfer, switching from an oxidizing form [NAD+] to a reducing form [NADH] and vice versa [29], it is involved in numerous metabolism pathways, including being a necessary co-enzyme for Sirt1 activity. Sirt1 regulates and controls many key transcription factors and co-factors involving in metabolic homeostasis; due to this significant downstream effects, Sirt1 is increasingly considered as a master regulator for metabolism [30]. Accordingly, Sirt1/SIRT1 has been widely investigated in the field of metabolism in different key organs and tissues including the liver, pancreas, muscle, adipose tissue (see Table 1), and brain. Specifically, SIRT1 is expressed in the hypothalamus, where it is distributed in the ventromedial, arcuate, paraventricular and dorsomedial nuclei, not only supporting its critical role in metabolic regulation in the brain [31] but also more broadly links Sirt1 to the hypothalamic–pituitary–gonadal and -adrenal axes, and thermoregulation, circadian rhythms and satiety, several of which we discuss later. Generally, disruption in energy metabolism is common among AD population such as eating behaviors [32]. Brain Aβ plaques and NFTs are common in AD’s hypothalamus and metabolic disorders such as obesity, insulin resistant, and diabetes worsen Aβ accumulation and tau tangles [33–35]. Unsurprisingly, Sirt1 is closely related to metabolic disorders including T2DM, obesity, dyslipidemia, and fatty liver disease [30]; all of which increase dementia risk.
The contribution of Sirt1 to metabolism in different organs and tissues
AMPK, Adenosine monophosphate-activated protein kinase; CRTC2, CREB (cAMP response element-binding protein) Regulated Transcription Coactivator 2; eNOS, endothelial nitric oxide synthase; FOXOs, Forkhead box O transcription factors; FXR, farnesoid X receptor; LKB1, Liver Kinase B1; LXR α, liver X receptor alpha; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; p53, tumour protein P53; PGAM-1, Phosphoglycerate Mutase 1; PPARα, Peroxisome proliferator-activated receptor (PPAR)-alpha; PPARγ; PPAR gamma; PGC-1α, PPARγ coactivator 1-alpha; prdm16, PR/SET Domain 16; SREBP1c, Sterol regulatory-element binding proteins 1c.
Sirt1 and cholesterol metabolism
Cholesterol homeostasis is necessary for brain development and neuronal functioning, so it is precisely regulated. Increased levels of circulating cholesterol have been associated with an elevated risk of dementia and AD, and several studies have found a reduced risk of AD in patients using cholesterol-lowering drugs [36]. Any cholesterol that is excess to the body’s need is oxidized into the oxysterols, 24-hydroxycholesterol (24-OHC) and 27-hydroxycholesterol (27-OHC), by the enzymes CYP46A1 and CYP27A1, respectively [37]. Both of these metabolites influence cholesterol biosynthesis in the brain [38], with 27–OHC suggested to elevate the risk of dementia and AD, while 24-OHC is suggested to have neuroprotective effects [37, 40]. While there is an efflux of 24–OHC from the brain to the peripheral circulation, there is also an influx of the neurotoxic 27–OHC to the brain [41, 42]. There is a close correlation between cholesterol and 27-OHC in the circulation and hypercholesterolemia is likely to result in an increased uptake of 27-OHC, which appears to mediate the effect of hypercholesterolemia; therefore, this sterol could be one of the primary pathogenetic marker for AD incidence [43].
A beneficial effect of 24-OHC might be through modulating the deacetylase activity of Sirt1, which in turn contributes to accumulation of AD pathological features. Specifically, a recent animal study in mice revealed that 24-OHC may act as a neuroprotective metabolite by potentiating Sirt1 activity, which in turn prevented the hyperphosphorylation of tau. The underlying mechanism for the protective effect of 24-OHC is proposed to be through improved Sirt1-dependent deacetylation, which prevents the neurotoxic degradation of p-tau, making p-tau more susceptible to ubiquitination and reducing tau tangles in neurons [40]. Sirt1 also plays a crucial role in cholesterol metabolism by deacetylating, and therefore activating LXRs, which are a key regulator of lipid metabolism [44]. The main function of LXRs include reverse hepatic cholesterol transport, regulating efflux of high density lipoprotein cholesterol-containing lipoproteins, degrading lipoproteins, and removing cholesterol from the body by conversion to bile acids [44]. A recent study showed that a mutation of LXRα at Lys-432 disrupts the deacetylation activity of Sirt1, and thus impaired LXR transcription, highlighting the importance of LXR deacetylation by Sirt1 [45].
Sirt1 and inflammation
Sirt1 is associated with many chronic inflammatory diseases including obesity, heart disease, and type 2 diabetes mellitus (T2DM), all of which are either common co-morbidities of, or major risk factors for, dementia and AD [46–48]. Evidence has emerged that Aβ accumulation is modulated by inflammation in brain cells, leading to neuronal loss and the progression of AD [49, 50]. The amyloidogenic pathway can stimulate inflammation processes and induce the brain’s immune system to release pro-inflammatory cytokines [51, 52]. Further, epidemiological studies have shown that non-steroidal anti-inflammatory drugs (NSAIDs) are associated with less cognitive deficits in older adults and reduced AD risk [53]. Sirt1 can reprogram inflammatory responses by influencing histones and transcription factors, such as NF-κB [54]. There is a strong association between inflammatory events in the immune system, metabolic pathways, and mitochondria to meet the nutritional requirements of the immune system. The sirtuin family appears to be involved in the regulation of these complex networks [54, 55]. NF-κB is considered to be a master inflammation regulator where, in response to pro-inflammatory cytokines, it translocates to the nucleus and induces the transcription of a cascade of pro-inflammatory cytokines and chemokines [56, 57]. Moreover, NF-κB may also mediate synaptic plasticity, learning, and memory [58]. A recent study has demonstrated impairment of NF-κB in AD associated with the characteristic reduction in ability to form new memories [58]. Sirt1 impacts NF-κB by deacetylation of p65 at lysine 310 [59], suppressing NF-κB transcriptional activation which is induced by the pro-inflammatory cytokine tumor necrosis factor alpha (TNFα). The inhibition of NF-κB signaling has been suggested to be a mechanism through which Sirt1 could protect neuronal cells against microglia-dependent Aβ toxicity [60]. Elevated Sirt1 levels could also suppress the inflammatory response in the hippocampus and thus augment cognitive abilities [61].
Other biological pathways relevant to AD
Circadian rhythm and the gut microbiome are two examples of AD-related biological processes that can be influenced by lifestyle [62, 63]. Moreover, growing evidence highlights the importance of Sirt1/SIRT1 for the regulation of circadian oscillation and gut microbial diversity [64, 65]. Here we summarize biological mechanisms that link these processes with Sirt1 and AD.
Sirt1 and circadian rhythm
Some behaviors follow a daily rhythm, including sleeping and eating [66]. The circadian clock is a complex biological system that enables adaption of human cells to 24-hour day and night cycles. The cellular circadian system comprises both a positive and negative limb. The positive limb is composed of BMAL1 (brain and muscle ARNT-like1) and CLOCK (Circadian Locomotor Output Cycles Kaput), while the negative limb is composed of two cryptochrome (CRY1 and CRY2) and three period (PER1, PER2, and PER3) proteins. These two limbs form a negative feedback loop where CLOCK and BMAL1 bind together and enhance the transcription of PER and CRY genes. The resultant PER and CRY proteins form complexes along with other proteins (PER complex) to suppress the activity of CLOCK-BMAL1 until the PERs and CRYs degrade [67]. The suprachiasmatic nucleus (SCN) in the hypothalamus regulates mammalian circadian timekeeping [68]. There is a reciprocal link between metabolic homeostasis, SCN and peripheral tissues regarding the circadian clock and neurological regulation [62, 69]. However, aging, along with features of modern life such as prolonged exposure to light at night and shift work, predispose humans to circadian dysfunction [70].
There is growing evidence to suggest that circadian dysfunction is both a cause and a consequence of neurodegeneration. Neurodegenerative diseases likely impair the central clock and disrupt the links between different brain regions and peripheral organs. On the other hand, disruptions to rhythmic process such as sleep and metabolism may increase the susceptibility of the brain to further degeneration [62]. Animal studies provide evidence that circadian dysfunction can contribute to AD pathology either directly, by exacerbating Aβ deposition, or indirectly via effects on the metabolic cycle [62]. Sirt1 is considered a potential regulator of circadian gene expression through PGC1-α, thereby modulating the transcription of core CLOCK genes, including CLOCK, BMAL1, PER2, CRY1, and ROR γ (retinoic acid-related orphan receptor gamma) [64]. In mice, aging was accompanied by decreased Sirt1 levels in the SCN and therefore a reduction in circadian regulatory proteins in the brain, highlighting the significant contribution of Sirt1 to age-related reduction in circadian function [71, 72]. The activity of Sirt1 also depends on the cellular level of NAD+ with production modulated by the circadian machinery, creating a critical link between the circadian clock, metabolism, and Sirt1 activation [10]. Therefore, it seems that any age-related impairment in the circadian system and Sirt1/SIRT1, in addition to that discussed above, might further impact metabolism and lead to health issues.
Sirt1 and gut microbiome
The gut microbiome, referred to as the ’forgotten organ’, comprises all microbes living in the gastrointestinal (GI) tract, including bacteria, viruses, fungi, and archaea [73, 74]. GI microbiota affect human health, either directly by generating biological substances such as essential amino acids, vitamins and lipids, or indirectly by mediating metabolism, intestinal epithelium barrier function, and immune responses [74]. Gut microbial diversity varies among individuals due to genetic and lifestyle factors [74], and typically declines with age [75, 76]. Improvements to detrimental lifestyle factors such as poor diet, suboptimal sleep, circadian rhythm disturbance, chronic noise, and sedentary behavior have been shown to improve and protect against age-related decline in gut microbial diversity [77]. Strong evidence for a genetic contribution to individual variation in the gut microbiome comes from twin studies, where the microbiome composition of monozygotic twins is more similar than that of dizygotic twins [78, 79]. A recent study found lower GI microbial diversity in AD patients compared to controls [63], with AD patients showing a higher frequency of Bacteroidetes, and reduced frequencies of Firmicutes and Bifidobacterium [63].
One possible explanation for the reduced number and diversity of gut microbes in people with AD, in addition to concomitant dietary-decline, is the reduction of their adhesion to the intestinal wall due to changes in the chemical composition and structure of the colon mucous membrane [80]. Alternations in gut microbiota play roles in disease progression, probably through influencing on immune system and systemic inflammation [81]. Although, the exact mechanisms which the gut impacts on neuropathology, emerging evidence propose the role of a gut-brain axis allowing bi-directional link between the gut and brain [82], both at the genetic [83] and microbial diversity level [84]. Sirt1 might impact gut microbiome homeostasis by improving intestinal barrier functions [85, 86], which may mechanistically result through Sirt1 deacetylation of SPDEF (SAM pointed domain containing ETS transcription factor), which is involved in the differentiation of the epithelial lining of the small intestine [87]. Other potential mechanisms by which Sirt1 may affect the gut microbiome include an anti-inflammatory effect of Sirt1 to reduce gut-related reactive oxygen species [65], its anti-apoptotic role to improve intestinal structure [86], and regulatory effects on tight junction-related genes [88].
SIRT1, LIFESTYLE FACTORS, AND AD COMORBIDITIES
A suboptimal lifestyle might be one of the main modifiable risk factors for global burden of chronic diseases and accounts for about 63% of all deaths [89]. There is growing evidence to suggest that addressing lifestyle factors such as, but not limited to, diet, physical activity, and sleep could be a viable preventative strategy for AD [90]. In the following sections, we summarize previous studies of potential associations between Sirt1/SIRT1 and lifestyle factors with respect to dementia and AD-related risk factors. Lifestyle factors that will be reviewed here in relation to Sirt1/SIRT1 and AD include dietary factors, physical activity, and smoking, while we have reviewed sleep previously [91]. There are several known common comorbidities linked with AD, with evidence at the mechanistic, epidemiological, and genetic level associating multiple disorders, such as those of the cardiovascular system, and gastrointestinal tract as well as T2DM and depression. In this review, we highlight the role of Sirt1 in depression, the most common psychiatric disease in the population and estimated to affect around 40% of people with AD.
Sirt1 and dietary factors
Caloric restriction, defined as up to a 50% reduction in calorie consumption, has been suggested to have significant neuroprotective effects [92] in addition to being associated with an increased lifespan, delayed aging, and protection from age-related diseases, such as AD [93, 94]. Young AD transgenic mouse models who were subject to caloric restriction displayed reduced accumulation of Aβ plaques and activation of astrocytes [95, 96]. This was associated with upregulation of genes linked to neurogenesis and neuroplasticity, and downregulation of inflammatory genes [97]. A recent study showed that habitually higher dietary energy intake results in an increased risk of AD incidence in individuals carrying the apolipoprotein E (APOE) ɛ4 allele [98], a major genetic risk factor for AD. Similarly, in individuals with mild cognitive impairment (which often but not always precedes dementia) who followed a high calorie diet, the risk of developing AD increased two fold compared with subjects following a lower calorie diet [99]. Recent research has shown that caloric restriction in transgenic mice could simulate SIRT1 overexpression, improving glucose metabolism and extending lifespan [100].
One of the best-described molecular mechanisms by which caloric restriction affects aging is via the Sirt1 signaling pathway (AMPK (Adenosine monophosphate-activated protein kinase)-Sirt1-PGC-1α) [101]. Both Sirt1 and AMPK are highly conserved energy sensors that respond to elevated levels of NAD+ and AMP, respectively. During fasting, elevated AMP/ATP (Adenosine triphosphate) ratio (an energy indicator) results in AMPK activation. AMPK directly phosphorylates PGC-1α, preparing this biomolecule to be deacetylated by Sirt1 and thus fully activated [102]. Sirt1’s deacetylation of PGC-1α has been extensively implicated in metabolic regulation and biogenesis of mitochondria and proposed as a potential mechanism linking Sirt1 to a role in caloric restriction and longevity [103]. Human studies have reported that DNA content in mitochondria increased by 35% in the skeletal muscle biopsies collected from participation with CR, demonstrating increased mitochondrial mass which is followed by increases in SIRT1 gene expression [104]. Despite these findings, significant further research is needed to clarify whether Sirt1 activation is a promising target to achieve good health and longevity in human being [105].
The Mediterranean diet (MeDi) is common in countries bordering the Mediterranean Sea [106]. It includes large quantities of fruits and vegetables, and omega-3 fatty acids from olive oil, which is believed to improve plasma antioxidant capacity [107]. Adherence to the MeDi has previously been linked with improved cognitive abilities, reduced inflammation, a lower risk of neurodegenerative diseases such as AD [108, 109], slower rate of brain Aβ accumulation, increased cortical thickness, and reduced brain atrophy [110] and improved longevity [111, 112]. It has been suggested that polyphenols in the MeDi have significant antioxidant properties, particularly resveratrol [113]. In an intervention study, it was shown that administration of resveratrol (10 mg/kg) over a period of 8 months could improve the integrity of the hippocampus and consequently cognitive performance in rats [114]. Resveratrol is a well-established activator of Sirt1 and appears to influence longevity through the activation of sirtuin pathways [115].
The Western diet, which is characterized by high intake of saturated fats and refined sugars and low intake of fiber, is a hallmark of the modern lifestyle and is emerging as a risk factor for AD and dementia [116]. Several studies have reported an association between the Western diet and various chronic diseases, including AD [116–118]. Both human and animal studies have shown that the Western diet can increase Aβ deposition and tau phosphorylation, and exacerbate cognitive decline, while following a diet lower in fat and sugar can improve dementia and cognition [119]. A proposed hypothesis is that oxidative stress is one of the primary consequences of a high energy intake and plays a critical role in cognitive impairment [120]. Sirt1 as a main regulator of metabolism modulates fatty acid metabolism via an interaction with several metabolic sensors, including AMPK and PGC-1α [121–123]. Other potential contributions of Sirt1 to fat metabolism include improving insulin signaling through fat mobilization and mammalian target of rapamycin signaling [124], and elevating insulin release in the pancreas, where it acts as an anti-inflammatory factor [125].
Sirt1 and physical activity
A sedentary lifestyle is a common feature of modern society and is a public health concern [126]. Being physically inactive has been shown to be a major risk factor for cognitive decline, dementia, and AD, while physical exercise appears to be an effective strategy to improve cognitive abilities [127, 128]. Skeletal muscle and brain are post-mitotic tissues and have limited ability for cellular renewal, making them particularly susceptible to age-related decline [129]. Both animal and human studies have shown that being physically active directly influences brain structure and function [130]. There is emerging evidence for neuroprotective effects of regular exercise, including improving oxidative stress status [131], elevating BDNF levels/expression [132], and mitochondrial biogenesis [133]. It is proposed that the beneficial effects of regular exercise on brain health and longevity could be due to Sirt1-related mechanisms.
Physical exercise causes cellular metabolic stress, which in turn impacts the Sirtuins. Metabolic stress increases ATP demand and leads to elevated levels of the Sirt1 substrate, NAD+. Consequently, elevated Sirt1 enzyme contributes to biogenesis of mitochondria through a PGC-1α-dependent pathway [134]. In a 6-week high intensity training study, an increase in mitochondrial activity and an elevation of PGC-1α protein was detected in skeletal muscle 4 days after training [135]. Sirt1 protein was also found to be decreased post-intervention while its total activity in muscles and activity per protein increased [135]. However, contradictory studies report that either Sirt1 protein increased after physical exercise [136], or that there were no reported changes in Sirt1 mRNA expression [137]. Of note, increased mRNA is not paralleled by increased protein because there are complex regulatory points for protein transcription after gene expression. Moreover, a recent study showed that 30 minutes post-exercise Sirt1 protein remained unchanged and its level increased after 120 minutes of recovery [136], therefore, sampling time point may impact outcome, as may the type of exercise prescribed and the cohort being investigated.
There is evidence to support the notion that exercise can improve learning processes and memory formation by increasing BDNF in the hippocampus [138]. Indeed, it seems that the important mechanism linking the neuroprotective effects of exercise to Sirt1/SIRT1 is mediated through BDNF [139], which plays a crucial role in neuronal plasticity and neuronal survival [140]. Sirt1 is thought to be a regulator of BDNF transcription [141], enhancing memory function and spatial learning. A recent animal study reported that the metabolite lactate, produced in muscles during exercise, might be a stimulus for BDNF and thus improved spatial learning and memory formation [138]. Importantly, the activity of lactate depends on Sirt1 deacetylase activity [138]. The most studied BDNF genetic polymorphism is rs6265, which results in a Methionine for Valine substitution at position 66 (Val66Met). The 66Met allele is associated with increased susceptibility to cognitive deficits and neuropsychiatric disorders [142]. Importantly, a recent study revealed significantly lower expression of vascular SIRT1 in transgenic mice homozygous for the 66Met allele [143]. Another study explored the possible influence of aging and moderate exercise on Sirt1 activity by measuring the acetylation rate of proteins in the cerebellum [129]. The authors found that Sirt1 levels did not change with age, but a positive association between hand grip strength and protein acetylation was reported, suggesting that age-related decline in Sirt1 activity in the cerebellum could damage motor function [129].
Sirt1 and smoking
It is estimated that around 2 billion people worldwide smoke tobacco, leading to around 4 million deaths annually [144]. Illnesses that have been linked to smoking include heart disease and cancer, along with a wide range of neurological disorders [145, 146] such as dementia and AD [147]. Previous studies have demonstrated that active or former smokers have a significantly higher risk for AD (relative risk = 1.72; 95% CI 1.33–2.12) [148], which is markedly higher in those who carry the APOE ɛ4 allele (OR = 6.56; 95% CI = 1.80–23.94) [149]. It has been proposed that cerebral oxidative stress is the main pathophysiological mechanism involved in the neurocognitive and neurobiological impairments observed in smokers [146]. The brain is a highly energy-demanding organ with a high rate of metabolism, producing a high burden of free radicals. Given brain tissue is rich in polyunsaturated fatty acids, it is highly susceptible to peroxidation and oxidative stress [150, 151].
Sirt1 has been reported to be reduced in smokers [152], who are characterized by an imbalance of oxidant/antioxidant capacity [153, 154]. In the presence of oxidative stress, Sirt1 shifts to the nucleus, to act as an antioxidant. It appears that under oxidative stress, Sirt1 can stimulate antioxidant enzymes such as Superoxide dismutase-2 and -3 (SOD2 and SOD3) by deacetylation of FOXO3a [155]. Evidence suggests that the anti-senescence pathway Sirt1/FOXO3 is impaired in smokers, leading to disruption of the antioxidant/pro-oxidant balance [156]. Further, Sirt1 protein has been suggested to protect endothelial cells against smoking–related oxidative stress. In an animal study, rats treated with cigarette smoke showed increased reactive oxygen species, inflammatory markers and induced apoptosis, in arteries and coronary arterial endothelial cells [157]. Following treatment with the Sirt1 activator resveratrol, antioxidant, anti-inflammatory, and anti-apoptotic effects were observed via Sirt1 activation [157]. It is hypothesized that oxidative stress due to smoking decreases SIRT1 expression and increases acetylated endothelial nitric oxide synthase, which is followed by endothelial dysfunction [158].
Sirt1 and depression
Depression is prevalent in older adults and is more common among people suffering from dementia, with a rate of 30% among AD patients [159]. Depressive symptoms are often detected before memory impairment [160] and could contribute to memory decline by slowing down the generation of nerve cells [161]. It has therefore been suggested that depressive symptoms could be an early sign of memory decline, and older adults who report early signs of depression may be at risk of developing dementia. [160]. Several animal and human studies have revealed a crucial role for Sirt1 and its activators in depression and other mood disorders [162, 163]. Neuroinflammation stimulates the brain’s immune system, including microglial activation and astrocyte hypertrophy, which are known to be key contributors to stress-induced depression [164]. Treatment with the Sirt1 activator resveratrol has been reported to attenuate microglia and astrocyte activation, and increase hippocampal neurogenesis in older rats, which improved depression-like behaviors [162, 163], highlighting Sirt1 as a potential target for future antidepressant drugs [51–53]. In a mouse model of depression, chronic stress interrupted Sirt1 activity in the hippocampus, resulting in depressive behaviors [165]. By contrast, increased Sirt1 activation improved depression-related behaviors [165]. Increased Sirt1-mediated phosphorylation of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) in the chronic stress state was proposed as the underlying mechanism, as activation and blocking of hippocampal ERK2 were followed by anti-depressive and pro-depressive behaviors, respectively [165]. Notably, mood impairments are also linked to changes in circadian rhythms, and circadian disorders in various physiological processes are reported in depression. As discussed earlier, there is emerging evidence to support an effect of Sirt1 on circadian rhythm by modulating the key circadian rhythm components of CLOCK and BMAL1 [166, 167].
SIRT
1 GENETIC VARIATION, AD, AND AD-RELATED RISK FACTORS
Single nucleotide polymorphisms (SNPs) in the SIRT1 gene, located on Chromosome 10 (10q21.3), have been associated with several AD-related risk factors, including cardiovascular disease and diabetes [168]. However, there is a lack of literature linking genetic variation in SIRT1 directly with AD. SIRT1 genetic variants reported by previous studies are summarized in Table 2, along with the diseases, disorders, or phenotypes they are associated with, many of which have themselves been reported as potential risk factors for AD. As such, we have reviewed these genetic associations to provide evidence to suggest that further investigation of SIRT1 genetic variation is warranted, particularly in the context of gene-environment (GxE) interactions and AD.
Association of SIRT1 genetic variants with diseases and traits related to aging and Alzheimer’s disease
SNP, single nucleotide polymorphism; NFLD, non-alcoholic fatty liver disease; COPD, chronic obstructive pulmonary disease; CVD, cardiovascular disease; T2DM, type 2 diabetes mellitus; BMI, body mass index.
As earlier mentioned SIRT1 has a crucial role in metabolism and metabolism-related disorders such as T2DM, which themselves have strong links or are considered as comorbidities or risk-factors for AD. As such, considering GWAS that have revealed an association of the SIRT1 genetic variants on metabolism, is of relevance to AD. Recent study examined SNP rs2273773 with different metabolic phenotypes, including metabolic syndrome, seasonal variation in body weight, fasting blood glucose, and energy expenditure [169]. In a Dutch study of 1,390 participants, an association between SIRT1 genetic variants and glucose tolerance was reported, which had implications for long-term survival. In this study, carriers of the minor allele at SIRT1 rs12778366 had higher glucose tolerance and reduced mortality risk compared with wild type subjects, and the protective effect of this variant was significant in overweight and obese participants [170]. A Finnish cohort study, which investigated three SIRT1 SNPs (rs3740051, rs2236319, and rs2272773) showed that the minor allele of these SNPs was strongly predictive of elevated basal energy expenditure in healthy subjects with diabetic parents [171].
With respect to the cardiovascular system and given that cardiovascular disease and other vascular factors are considered as significant risk factors for AD, the role of Sirt1/SIRT1 is important to consider. It has been shown that Sirt1/SIRT1 protects cardiomyocytes and endothelial cells against stresses and improves survival at the systemic level [172]. At the genetic level, in a recent study among 1,705 men aged over 70 years, SIRT1 SNPs and serum Sirt1 protein were evaluated and reported two SNPs (rs3740051 and rs3758391) to be associated with arthritis, heart attack, and deafness [173]. Moreover, analysis for association of SIRT1 SNPs with cognitive abilities revealed that rs2273773-C homozygotes had a lower score on the Mini-Mental State Examination (MMSE; measure of global cognitive function) than rs2273773-T homozygotes [173]. Similarly, in a longitudinal investigation of 4,573 Rotterdam Study participants, variation at three SIRT1 SNPs (rs7895833, rs1467568, and rs497849) was evaluated against all-cause mortality [174]. While there was no association between SIRT1 genetic variation and all-cause mortality in the whole population, diabetic patients carrying risk variants had 50% higher risk of all-cause mortality, especially cardiovascular mortality. This risk was greater in smokers and individuals with lower intake of the water-soluble B vitamin, niacin [174], the former reiterating the link between Sirt1 and smoking discussed previously.
Recently, it has been reported that increased Sirt1 levels in the brain, particularly in the lateral hypothalamic and the dorsomedial nuclei, slows down aging and increases longevity in mice [175]. Several GWAS have also assessed SIRT1 SNPs in relation to lifespan and longevity, though there is a lack of consensus in the findings. In a Chinese case-control study of longevity, including 500 individuals, the SIRT1 rs4746720 variant was reported to be associated with human longevity [176]. Similarly, Kim et al. evaluated the allele and genotype frequencies of SIRT1 rs7896005 in Caucasian subjects. The authors found that the frequency of the minor allele of rs7896005 was associated with greater mean longevity [177]. However, a further study including 616 long-lived Chinese (age: 102.4±2.3 years) and 846 controls (age: 48.9±10.6 years) showed no association between 8 SIRT1 SNPs and human lifespan, at the allele, genotype or haplotype level [178].
While genetic variation in SIRT1 appears to be involved in the incidence and progression of diseases and phenotypes that themselves are risk factors for AD, there is a lack of literature directly providing evidence for associations of SIRT1 variation with either AD risk or its related pathological phenotypes, with the exception of the association of SIRT1 rs2273773-C homozygotes with lower MMSE score [173]. This raises the possibility that Sirt1/SIRT1 may impact AD in a more complex manner, potentially via interaction with environmental or lifestyle factors (i.e., GxE). As discussed earlier, one such group of factors that has gained considerable attention in AD is that of modifiable lifestyle factors or behaviors, including diet, physical activity, and sleep. These factors also offer promise as the foundation of intervention strategies aimed at delaying the onset of, or even preventing, AD. In the following section, we consider how Sirt1/SIRT1 may alter the effects of such modifiable lifestyle factors and behaviors.
SIRT1 genetic variation and lifestyle interactions
Studies of DNA sequence variation are enabling progress towards personalized medicine, which aims to determine an individual’s predisposition to various diseases as well as predict their likely response to external factors such as different nutrients, drugs, pathogens, and chemicals. While these approaches, when focused on pharmacological responses, can provide opportunities to target specific treatments to an individual to achieve the greatest efficacy and/or avoid potential side-effects, when focused on genetics and environmental factors such as lifestyle, personalized medicine can contribute to identifying the most effective non-pharmacological intervention strategy for the individual [179].
Currently there is a lack of studies investigating whether genetic variation in SIRT1 can modify the effect of lifestyle factors (e.g., diet, sleep, and physical activity) on dementia and AD development. However, there is some evidence that interactions between SIRT1 gene variations and lifestyle factors can affect outcomes of other chronic diseases, some of which are themselves AD risk factors. In addition to the investigation in the Rotterdam Study [174] discussed above, which reported on the impact of SIRT1 interactions with smoking on all-cause mortality, a further study by Weyrich et al. investigated whether SIRT1 genetic variation could explain differences in the effect of a 9-month-long lifestyle intervention (including caloric restriction and increased physical activity) on metabolic outcomes in 196 participants of the Tubingen Family Study who were at high risk of diabetes [48]. At baseline, carriers of the minor allele (A) of SIRT1 rs12413112 had significantly lower basal energy expenditure and a higher respiratory quotient than homozygotes for the major allele (G). Participants carrying the rs12413112-A allele were also poorer responders to the lifestyle intervention than GG homozygotes, showing less improvement in fasting glucose and insulin sensitivity, and a much smaller reduction in liver fat content [48], all factors associated with increased AD risk. These findings provide impetus for future studies that should be aimed at exploring the interaction between genetic variation in SIRT1 and lifestyle factors, such as diet, sleep, and physical activity, and how this interaction may impact AD risk and pathophysiology.
FUTURE RESEARCH DIRECTIONS
This review has summarized the growing body of literature, from both in vitro and in vivo (animal and human) studies, that evidences a multi-factorial relationship between Sirt1/SIRT1, and AD etiology. This evidence supports a direct relationship with AD, but also indirectly through associations with AD related biological pathways and comorbidities, both at the functional and genetic level. There is also developing evidence that this relationship is complex and modulated through the impact of environmental and lifestyle factors. However, further research is required to fully elucidate this relationship, particularly as the direct relationship of SIRT1 gene-lifestyle interactions with AD has not received significant attention. Several lines of further investigation are thus recommended. One obvious recommendation for future research is to comprehensively examine genetic variation in SIRT1 in relation to AD-related phenotypes (i.e., cognition and brain-imaging) in comprehensively phenotyped cohorts. This should be done both independently and as an interaction with modifiable lifestyle factors (e.g., physical activity, sleep, and diet) to provide more conclusive evidence as to whether there is a either a direct genetic relationship or whether SIRT1 genetic variation may moderate lifestyle associations. This could then be extended to the epigenetic level, to determine whether this is a potential mechanism through which lifestyle factors influence AD-related phenotypes. While observational studies could provide significant evidence to support the relationship between Sirt1/SIRT1 in AD, cohorts in which lifestyle interventions have been undertaken could also provide an indication to any mechanisms underpinning associations (e.g., epigenetic, gene expression) and/or whether SIRT1 genetic variation impacts the efficacy of such interventions. Taken together, this evidence could provide support for targeting specific lifestyle interventions to specific groups of individuals based on underlying genetics. Further, it could also provide evidence to support the investigation of activators of Sirt1, e.g., dietary approaches (e.g., caloric restriction), resveratrol, etc., and their efficacy for delaying or potentially even preventing the onset of dementia due to AD.
CONCLUSION
Across this review, we have presented several lines of functional evidence that Sirt1 is associated with various biological processes that are relevant to AD, including Aβ and tau metabolism, cellular and cholesterol metabolism, inflammation, and circadian rhythm. We have also summarized literature that suggests environmental factors, particularly lifestyle factors, directly impact Sirt1 levels and/or function. Further, while there is a lack of literature directly linking SIRT1 genetic variation with AD, there is evidence that SIRT1 variation is directly linked to diseases and phenotypes that themselves are risk factors for AD. There is also evidence, albeit limited, that SIRT1 genetic variation modifies the impact of lifestyle factors on chronic disease processes which are linked to AD risk. When considering this multifactorial evidence collectively, we conclude that the interaction of SIRT1 genotype with lifestyle represents a promising, yet significantly under-investigated, target for furthering our understanding of AD risk and progression. Further research in this area is warranted to aid in the development of potential personalized lifestyle intervention strategies for AD.
Footnotes
ACKNOWLEDGMENTS
Figures 1 ![]() were created with BioRender.com. The authors have no further acknowledgments to report relating to this manuscript.
were created with BioRender.com. The authors have no further acknowledgments to report relating to this manuscript.
FUNDING
MM is supported by an Edith Cowan University Higher Degree by Research scholarship. TP is supported by an Edith Cowan University Strategic Research Fellowship. SRRS is supported by a National Health and Medical Research Council (NHMRC) Emerging Leadership Investigator Grant (GNT1197315). The authors have no other funding to report that is relevant to this manuscript.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.