
Abstract
Alzheimer’s disease (AD) is the primary cause of dementia affecting millions each year across the world, though still remains incurable. This might be attributed to the lack of knowledge about the associated proteins, their cellular and molecular mechanisms, and the genesis of the disease. The discovery of drugs that earlier revolved around targeting the amyloid-β cascade has now been reformed with the upgraded knowledge of the cross-seeding ability of tau protein which opens new gateways for therapeutic targets. This article provides a comprehensive review of various direct and indirect connecting pathways between the two main proteins involved in development and progression of AD, enabling us to further expand our repertoire of information regarding the etiology of AD. The current review indicates the need for extensive research in this niche, thus considerable advances can be made in understanding AD which eventually helps to improve the current therapeutics against AD.
INTRODUCTION
Alzheimer’s disease, named after its discoverer Dr. Alois Alzheimer, is a neurodegenerative disorder characterized by the formation of extracellular insoluble plaques of amyloid-β (Aβ) protein and intraneuronal tangles of tau protein (τP) developed in the neuronal cells [1]. Amyloid-β protein precursor (AβPP) is a transmembrane protein of neurons which is involved in the growth and repair mechanism [2]. These proteins are cleaved by two enzymes, α-secretase and γ-secretase, producing a soluble peptide which will be degraded; however, in the presence of another enzyme, β-secretase, along with γ-secretase, it produces an insoluble sticky Aβ monomer which aggregates outside neuronal cells [3, 4]. The plaques formed are localized between synapses disrupting signals and further affecting memory [5, 6]. Neurons are held together by cytoskeleton made up of microtubules, which are bound together with the help of τP which when phosphorylated tend to change their shape and cluster together forming neurofibrillary tangles inside the cells [7–9], driving it toward apoptosis [10]. The symptoms begin with short term memory loss, then as the accumulation of proteins proceed, the loss of motor skills and language are also observed [11, 12].
There exist two classes of AD, namely sporadic (late-onset) AD and familial (early-onset) AD [13]. The manifestation of the former one is associated with genetic and environmental factors, which is also affected by the succession of age [14]. An apolipoprotein (APOE) gene, which is affected, whose product binds to Aβ polypeptide, plays a major role [15]. This gene has 3 major alleles (E2-E4) among which the less effective one is E4 and therefore its presence makes the individual more susceptible to acquiring AD [16]. In familial AD, genes that happen to be mutated are presenilin 1 (PSEN1) and presenilin 2 (PSEN2), which constitute the catalytic subunits of γ-secretase or APP gene, resulting in the accumulation of Aβ polypeptides [17].
Aggregation of Aβ and τP is invariably observed in both forms of AD, which drives the downfall of the central nervous system and into developing the characteristic symptoms of AD.
This is already an established theory that aggregation of Aβ proteins starts about 20 years [18] and that of tau about 15 years prior to the occurrence of the first observed symptom, indicating a possible connection between the two proteins [19]. However, the already present Aβ protein has been found to induce the phosphorylation of τP proteins [20, 21], further causing their detachment from the microtubules of the axon and forming tangles. In the race of discovering a cure to AD, the scientists have targeted Aβ peptide to prevent its aggregation in the neuronal cells; however the outcome of clinical trials carried out for these drugs have not been promising [14]. Presently, scientists have changed their focus from targeting Aβ plaque aggregates to the interaction between the two proteins, the role it plays in inducing phosphorylation of tau, in order to reduce the rate of deterioration of neuronal cells. On the other hand, some studies have also revealed the role of τP in inducing Aβ toxicity in cells [22], generating a vicious cycle of pathogenesis, which gets established in AD.
Here in this review, we discuss the direct and indirect interacting models of Aβ and τP proteins that have been put forward by various researchers, to understand the feedback loop of the two proteins involved, and, moreover, continuously explore new potential targets for therapeutic drugs.
DIRECT INTERACTIVE PATHOGENESIS
Neurodegenerative diseases like AD and Parkinson’s disease, where aggregation of prion-like proteins is caused due to cross-seeding, have shown to produce prominent pathological implications [23]. In vitro studies have presented with evidence of the direct interaction between Aβ and τP proteins forming stable insoluble forms, further inducing toxicity. The strength of association to Aβ protein is dictated by the nature of τP, i.e., whether it is in phosphorylated or dephosphorylated form [24]. The phosphorylation of serine and threonine amino acid residues of τP has been shown to decrease its affinity toward Aβ protein, with the help of techniques such as Enzyme linked immunosorbent assay and western blotting [25]. This dependency indicated the direct role of Aβ in the phosphorylation of τP proteins, and alongside, in vivo demonstration of the co-localization of proteins was displayed by immunofluorescence assay [25].
Moreover, the researchers were able to discover the interactive sites on these proteins by utilizing peptide membrane arrays, exons 7 and 9 of τP, and middle to c-terminal region of Aβ were discovered to be the interacting sites [25]. This common interactive site occurs to be exploited in identifying a therapeutic drug target to reduce prion aggregation [26].
INDIRECT INTERACTIVE MODELS
Here presented are the brief outlines of various models describing the indirect interactions between Aβ and τP proteins that invariably occur through cell signaling pathways.
GSK3: One of the culprits
Glycogen synthase kinase 3 (GSK3) phosphorylates tau protein [27], which causes its detachment from microtubules and then their aggregation. The Aβ plaques hinder both insulin [28] and Wnt signaling pathways [29], creating an abundance of activated GSK3 in the neuronal cells. In the insulin pathway, Aβ acts as an antagonist to insulin, inhibiting phosphoinositide 3-kinase (PI3), and Akt (serine/threonine-specific protein kinase) activation, as shown in Fig. 1. Akt kinase phosphorylates two isoforms of GSK3, i.e., GSK3α and GSK3β, causing it to lose its kinase property and to be able to phosphorylate τP. However, if Akt is inhibited, the GSK3 is not phosphorylated and is available with its active phosphorylating property [28].
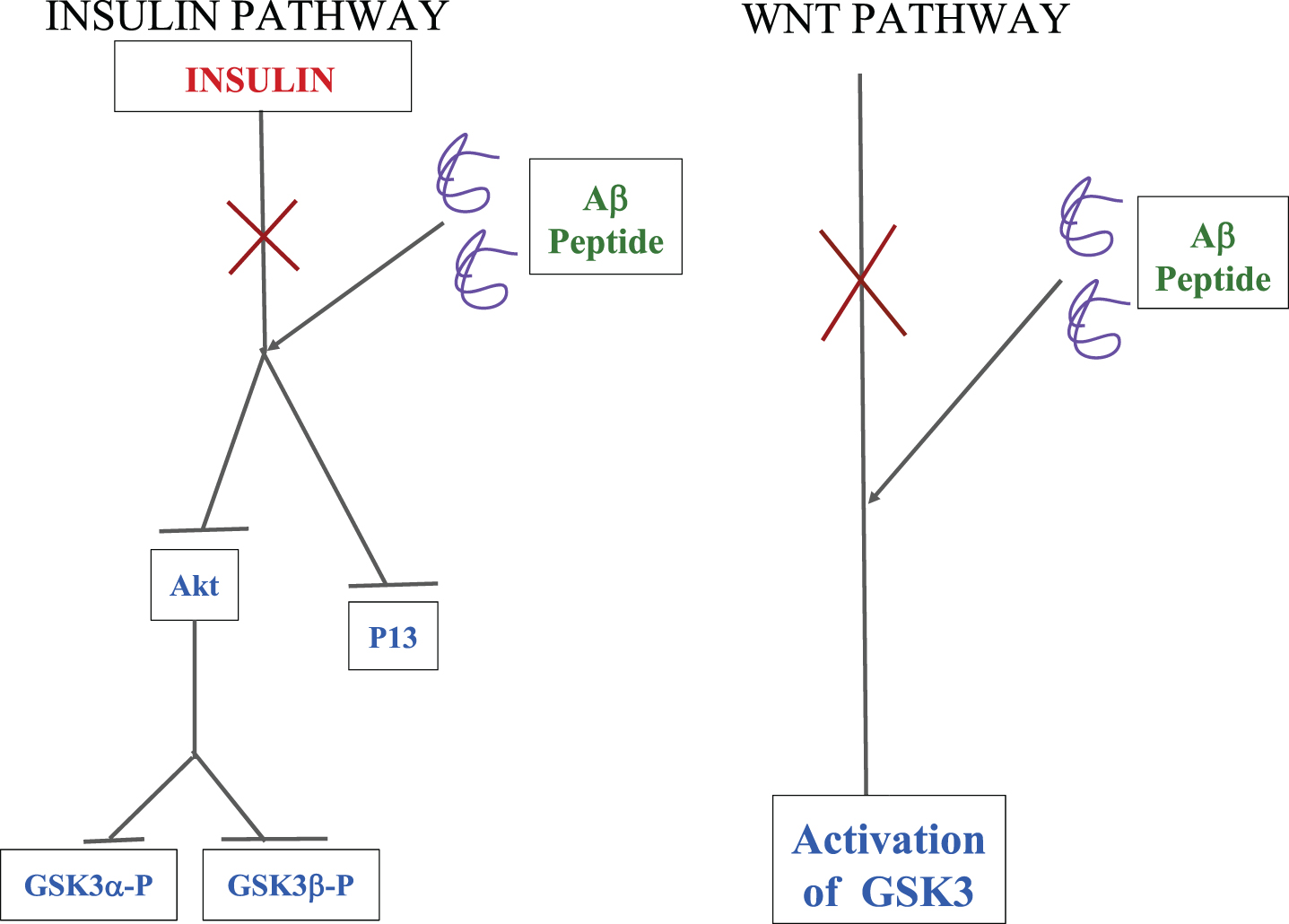
A schematic representation of how Aβ oligomers interfere with the insulin and Wnt pathways, leading to an increase in the levels of activated GSK3 in the cell which ultimately leads to neuron degeneration [30].
The Wnt pathway in one of its parts is responsible for the inactivation of GSK3; on the other hand, the presence of Aβ protein does not allow it to do that, and hence activated GSK3 peptides are present in the neuronal cells. The above-stated deviations of the cellular mechanisms are known to occur in familial AD [30].
CDK5: Another culprit
Cyclin-dependent kinase 5 (CDK5), a known kinase of tau protein, structurally similar to another kinase GSK3, is involved in regulating the cell cycle and several different cellular mechanisms in neurons [31]. The p35 and p39 forms are the activators of CDK5, which are involved in the proteasome degradation of CDK5, upon phosphorylation [32]; however, it gets cleaved into more stable p25 and p29 by calcium-dependent protease calpain. It was activated in presence of a high concentration of Ca2 + ions in the cell, an outcome of Aβ toxicity [33]. Hence, in the presence of stable activators, CDK5 remains active for a prolonged time in the cell causing phosphorylation of τP proteins [33] as shown in Fig. 2.
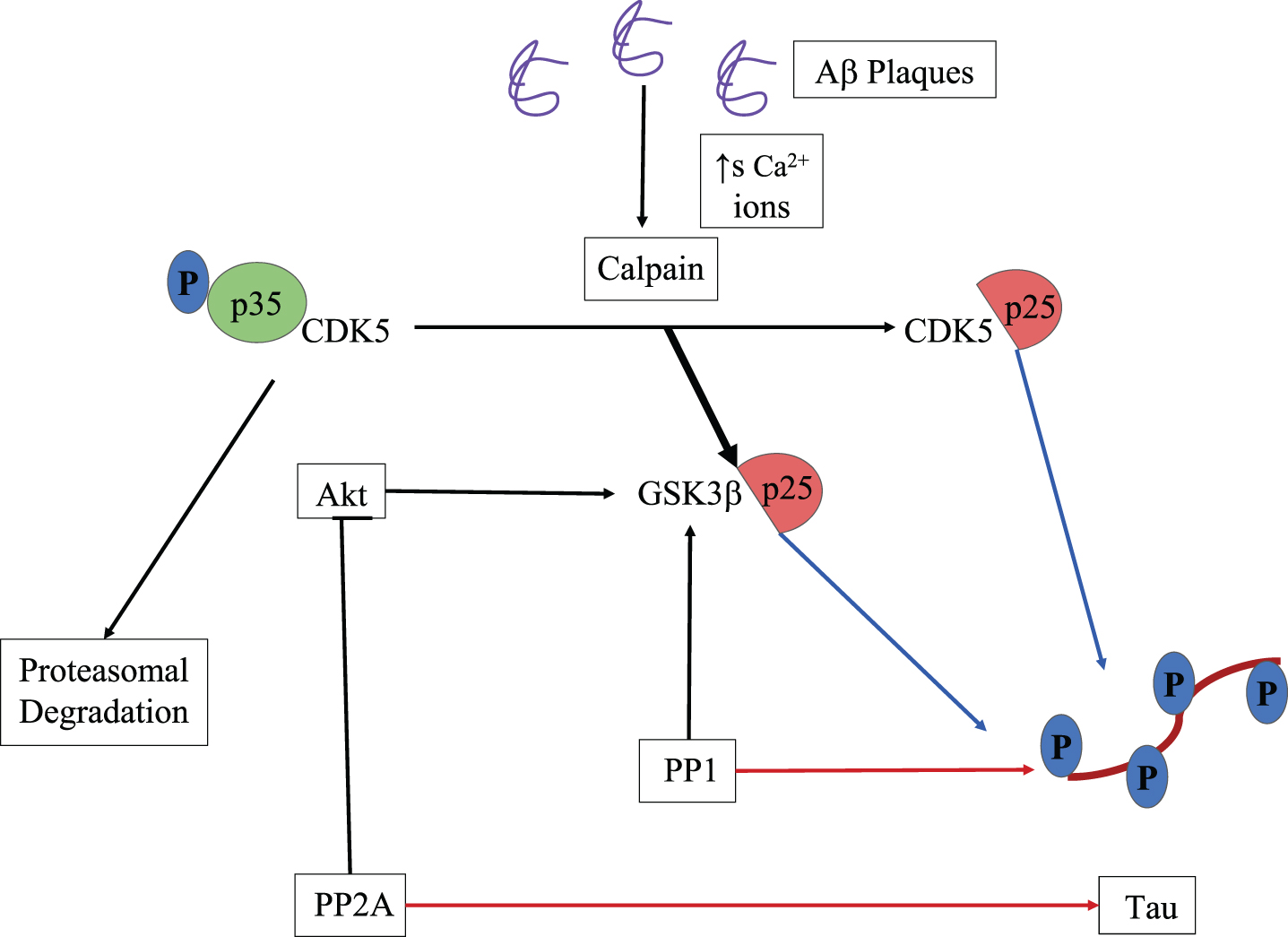
A schematic representation of both CDK5 and GSK3β induced phosphorylation of tau protein, activated by calpain cleaved p25. Protein phosphatases 1 and 2A which dephosphorylates tau protein also activate GSK3β leading to more phosphorylation than dephosphorylation. Thick black arrow indicates more affinity of p25 toward GSK3β than CDK5, blue and red arrows indicate phosphorylation and dephosphorylation activity respectively [33].
Protein phosphatases 1 and 2A are the main dephosphorylating agents of tau protein [34]. Protein phosphatases 1 is involved in activating GSK3β [35] and protein phosphatases 2A dephosphorylates Akt and thus inhibiting its activity [36]. The p25 has been shown to have a higher affinity toward GSK3β than CDK5, and therefore it binds to its C-terminus domain, causing an alternation in its activity, leading to an increase in phosphorylation of tau proteins [37].
Oxidative stress-mediated tau phosphorylation
Aβ oligomers interact with the neuronal membrane through receptors such as N-methyl-D-aspartate receptor (NMDAR), Ephrin type-A receptor 4 precursor [38], cellular prion protein [39], or can even get embedded into the lipid bilayer, leading to peroxidation of bilayer molecules that causing the damage to proteins, and hence taking our prime defense down [40].
Inside the cell, Aβ can induce tau phosphorylation through a plethora of pathways, for some of them the working mechanism is known (Fig. 3)
AGE-RAGE Stress [41] - Advanced glycation end products (AGE) and its receptor (RAGE) play a role in the etiology of AD. RAGE is a multi-ligand, and once Aβ transacts, it induces activation of Nuclear factor Kappa B (NF-κB), which further upregulates the level of various cytokines in the cell, and thus cause cell death [42]. Its activation also increases the expression of RAGE and makes cells more prone to external stress [42]. Their transact also activates NADPH oxidase [43], which leads to an increase in the concentration of reactive oxygen species (ROS) [44]. Cytokines are also known to upregulate this oxidase [45]. ROS further increases the production of Aβ oligomers [46]. Aβ hinders the activity of both complex I and IV of the electron transport chain [47] in mitochondria, by directly associating with subunit 1 of cytochrome c oxidase, the major site for the production of ROS and hence causing its upregulation in the cell [40]. Aβ induces higher expression of Regulator of calcineurin gene (RCAN1) through oxidative stress as it responds to it [48], which further inhibits the phosphatase activity of calcineurin, an enzyme that dephosphorylates tau protein. It also increases levels of GSK3β, a kinase known to phosphorylate tau [49]. The presence of Aβ has also been shown to decrease the levels of superoxide dismutase 2 (SOD2), which provide protection against the production of ROS, especially superoxides, and such a downregulation leads to their accumulation and increases in phosphorylation of τP. A plausible explanation can be due to the depletion of microtubule dependent protein 2 (MAP2), a protein required for the association of tau with microtubules, whose reduction has been linked to hyperphosphorylation of tau [50].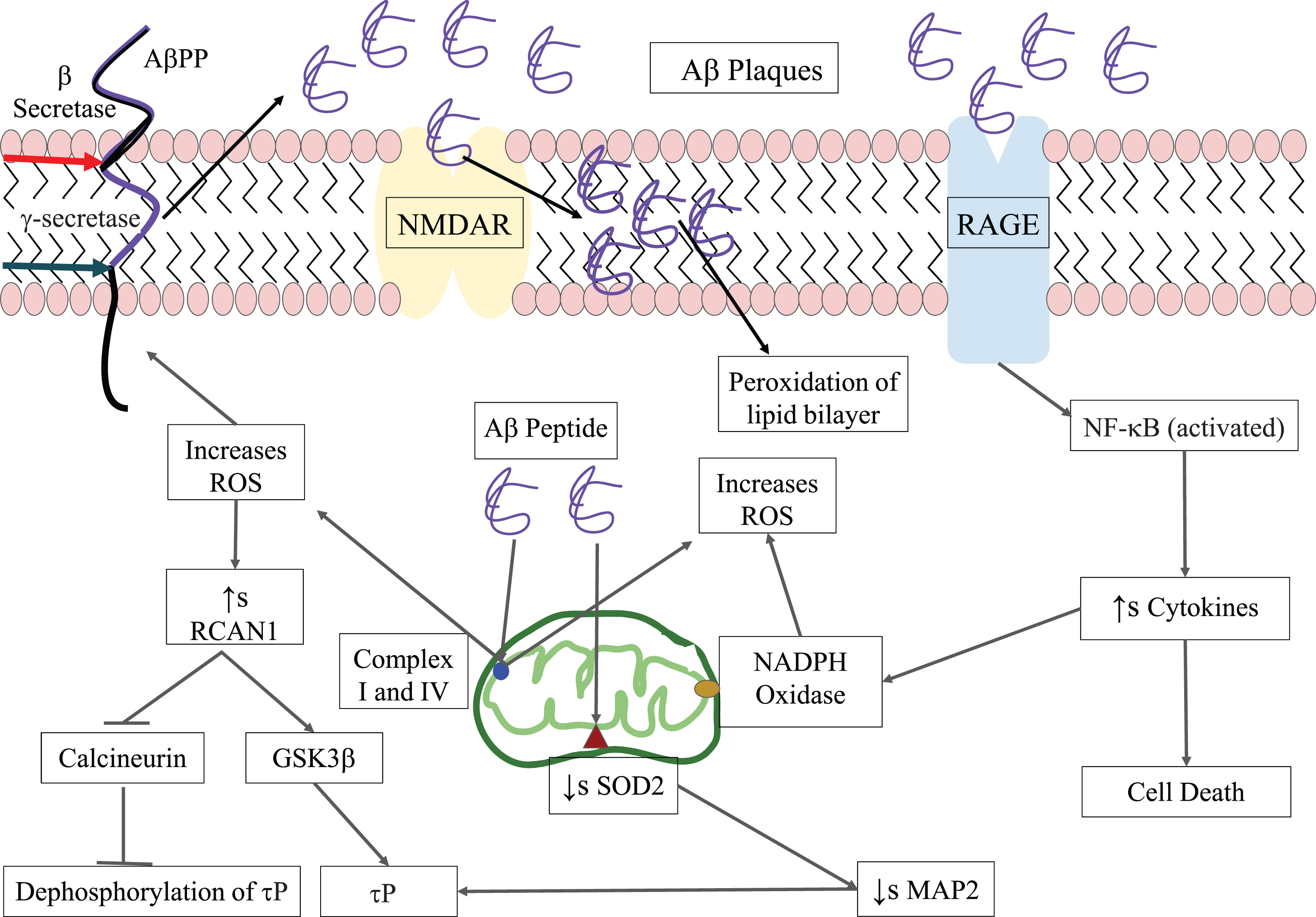
Acetylation of tau: associate of phosphorylation
Tau protein, similar to any other protein, can undergo various post-translational modifications including phosphorylation, acetylation [51], nitration [52], glycation [53], etc.
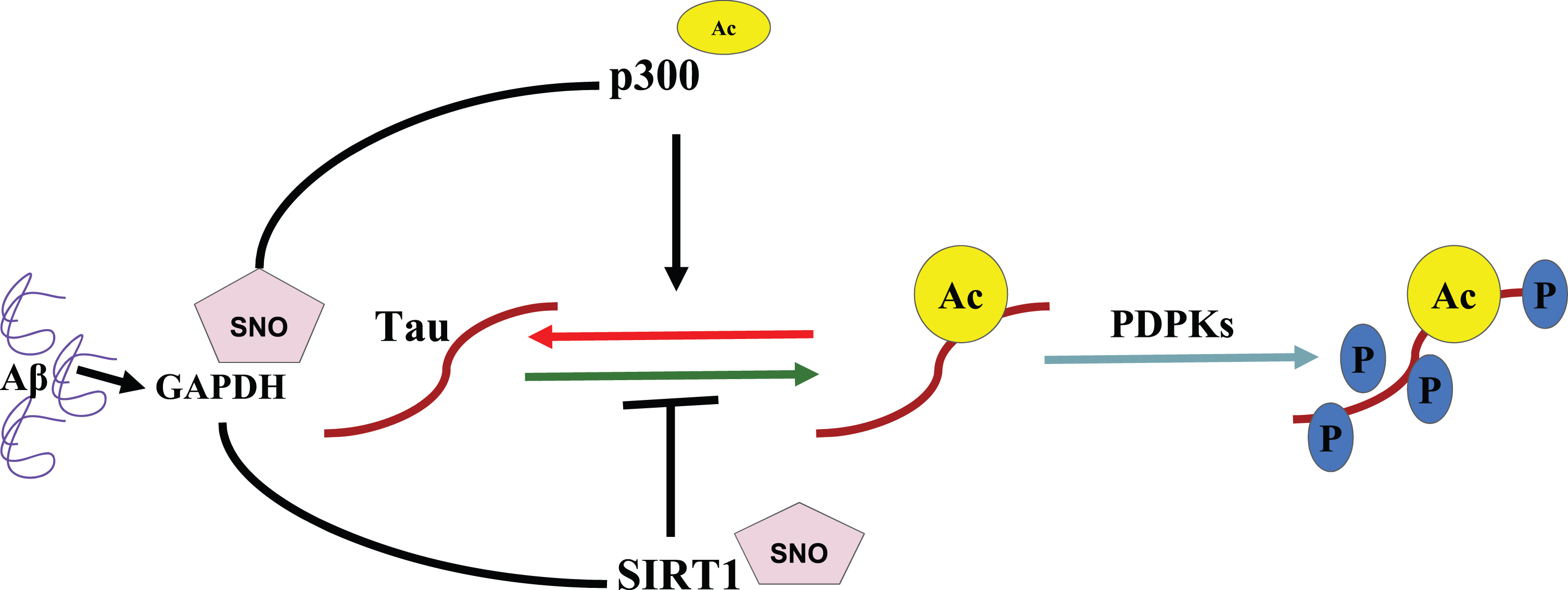
Aβ oligomers have been shown to induce nitric oxide (NO) production, nitrosylated glyceraldehyde 3-phosphate dehydrogenase (GAPDH) which acetylates p300 acetyltransferase, causing its activation and then acetylation of tau protein [54] as shown in Fig. 4. Nitrosylated GAPDH causes deactivation by nitrosylation of Sirtuin1 (SIRT1), a deacetylase of tau, further increasing the possibility of tau acetylation [55]. Its acetylation sets off its aggregation through phosphorylation by proline-directed protein kinases (PDPKs) [56].
A Schematic representation of how Aβ dictates tau phosphorylation via a GAPDH nitrosylated dictated pathway which acetylates p300 and nitrosylates SIRT1 promoting tau acetylation which furthermore causes its phosphorylation by PDKs [55].
Dendritic tau-dependent Aβ toxicity
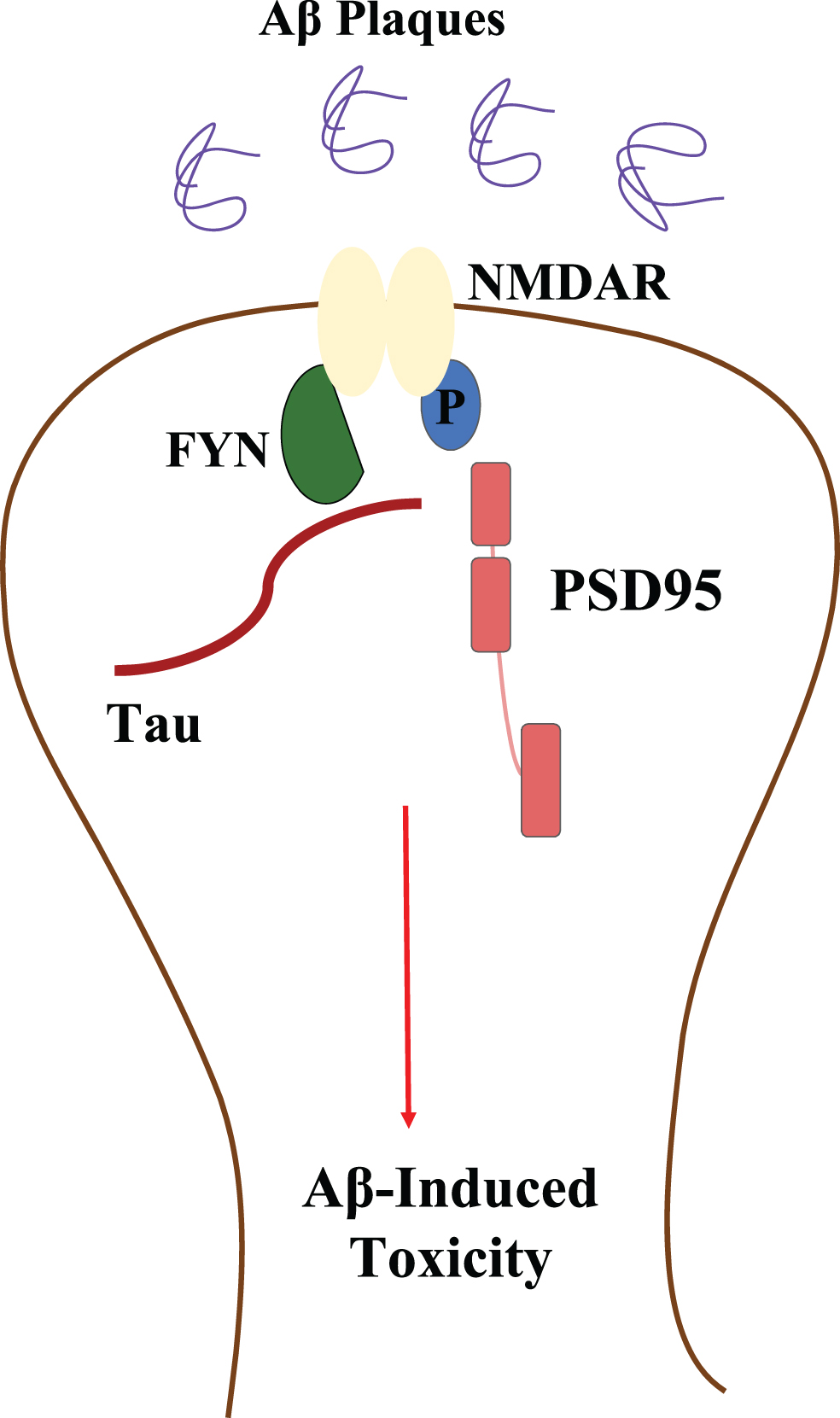
Tau protein, abundantly seen in axon and comparatively less in dendrites [57], upon phosphorylation, detaches from the microtubule and forms intracellular neurofibrillary tangles. These tangles of tau protein show greater affinity towards FYN, a tyrosine-protein kinase that is then localized to the somatodendritic region, further to phosphorylate NMDAR receptor present in the neuronal membrane and facilitates its interaction with postsynaptic density protein (PSD95) [58] (Fig. 5). Once NMDAR receptors are phosphorylated, they become more sensitive to the already existing Aβ plaques, which induce further downstream toxicity effects [22], and it also induces an influx of Ca2 + ions and affects other pathways, ultimately leading to neuronal loss [59].
A schematic representation of tau influenced Aβ toxicity by enhancing the sensitivity of NMDAR receptor to Aβ peptides through its phosphorylation by FYN kinase [58].
CONCLUSIONS
The interacting models of Aβ and τP stated in this review show the diverse signaling pathways that are involved in the etiology of AD, revealing the potential drug targets for the prevention or cure of the disease. Apart, there is a need to discover the other AD-associated pathways for a better understanding of the disease.
Earlier drugs used targeted the degradation of Aβ aggregates and failed to provide the desired outcome due to the seeding phenomena associated with the prion-like proteins. Nevertheless, the already phosphorylated tau proteins are sufficient to spread and cause toxicity to the cellular systems. The same situation persisted when τP tangles were targeted. Hence, aiming at only one of the proteins might not provide a cure to AD.
One current study that targets a common epitope involved in cross-seeding of the proteins with peptide-based inhibitors has shown effective results in blocking the pathological forms of Aβ and τP [26]. However, better inhibitors could be used for effective and prolonged inhibition of protein aggregations.
Designing a drug that helps cure AD or at least control its progression would require a complete understanding of the crosstalk between Aβ and τP, molecular mechanisms of other associated signaling cascade proteins, taking into account the pharmacodynamics and pharmacokinetics of the drug. The application of nanotechnology toward the bioavailability of drugs and enhancing its qualities and reducing any downstream toxicity can also be utilized.
Footnotes
ACKNOWLEDGMENTS
PK is thankful to the organizers of the Virtual Internship with Science Leader (#VISL), outcome which is this review article. SJM and SKP thank JSS Academy of Higher Education and Research (JSSAHER), Mysuru, Karnataka, India, for the infrastructure and support provided.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.