
Abstract
Neurocognitive disorders, such as Alzheimer’s disease (AD), affect millions of people worldwide and are characterized by cognitive decline. Human and animal studies have shown that chronic immune response and inflammation are important factors in the pathogenesis of AD. Chronic inflammation can accelerate the aggregation of amyloid-β peptides and later hyperphosphorylation of tau proteins. The exact etiology of AD is not clear, but genetics and environmental factors, such as age, family history, and lifestyle are linked to neurodegenerative diseases. Lifestyle habits, such as poor diet, are associated with inflammation and could accelerate or slow down the progression of neurodegenerative diseases. Here we provide a review of the potential conditions and factors that stimulate the inflammatory processes in AD. An understanding of inflammatory mechanisms influencing the development of AD may help to protect against dementia and AD.
INTRODUCTION
Due to a globally aging population, the numbers of those living with dementia are expected to grow threefold by 2050 [1], threatening a global dementia epidemic [2]. Alzheimer’s disease (AD) is one of the most common types of dementia. The estimated annual costs of caring for AD and dementia patients will increase from $307 billion to $1.5 trillion. A delay of AD onset by 1 to 5 years results in an economic gain of $183,227 to $511,208 [3]. In addition to economic gain, delaying AD is beneficial to individual and public health.
AD is a slowly progressing neurodegenerative disorder that is characterized by plaques of amyloid-β (Aβ) and neurofibrillary tangles which build up in the brain years or even decades before symptoms appear. Brain inflammation associated with aging may lead to increased amyloid plaque and tau tangle production and promote AD. The discovery of inflammatory pathways in the brain has led some researchers to propose that immunological/inflammatory mechanisms play a fundamental role in the development of AD [4–6]. Evidence suggests that AD patients experience chronic inflammation that is both driven by and enhances the dysregulation of the immune system.
Inflammation caused by disease states, such as obesity, which results in inflammation in the body’s periphery, can ultimately lead to the onset of neuroinflammation [4, 6]. It was once thought that the blood-brain barrier (BBB) insulated the brain from inflammatory insults to the periphery. However, it is now understood that a system of bidirectional communication exists between the peripheral immune system and the brain immune system [7]. In this way, increases in peripheral immune function can upregulate the activity of microglia and astrocytes. Furthermore, during the AD disease state, the BBB becomes leaky due to the accumulation of Aβ peptides around blood vessels [7–12], which may allow inflammatory mediators to infiltrate the central nervous system (CNS) from the periphery. During times of peripheral duress, the cytokines tumor necrosis factor-α and interleukin-6 can cross the compromised BBB and activate the central innate immune response, resulting in neuroinflammation and increased risk for AD [4, 13]. Peripheral inflammatory factors can activate immune cells such as microglia and astrocytes in the CNS, which are the major source of cytokines in AD [4, 14]. Prolonged neuroinflammation may contribute to the pathogenesis of AD [4, 15]. Therefore, appropriate treatment of chronic inflammatory conditions helps to prevent the disease.
The impact of dietary factors such as consumption of carbohydrates, vegetable oils rich in omega-6 fatty acids, and omega-3 fatty acids can have major effects on immune response and inflammation [16–18]. Diet and food-related conditions can cause chronic inflammation, which increases the risk of dementia and AD. A lifestyle that includes a healthy diet and physical activity can protect against cognitive decline and delay the onset of AD [16, 19]. In this review, diet and food-related conditions which can cause chronic inflammation that can be linked to AD will be discussed based on their main biological mechanisms of action. The purpose of this review paper is to explore the link between diet and inflammation and its effect on cognitive condition. For data gathering purposes, the PRISMA guideline was used. The initial search strategy identified some n = 1,426 papers; n = 120 studies were included after the abstract’s screening and n = 47 articles met the inclusion criteria. A new insight into how lifestyle habits, such as diet, fit into AD pathology through chronic inflammatory response may help to design new strategies to delay the progression of the disease.
VITAMINS
Vitamin B
The B vitamins are a group of eight micronutrients that serve as coenzymes in many important biological processes. The specific B vitamins B2, B6, B12, and folate are involved in recycling the amino acid homocysteine during the cyclic generation of the amino acid methionine. Deficiencies in the amounts of these B vitamins can lead to increases in plasma homocysteine levels [20]. Homocysteine facilitates the generation of reactive oxygen species (ROS), promotes oxidative stress, and increases inflammation [20, 21]. High levels of homocysteine is one of the risk factors for AD [22]. Vitamins B2, B6, and B12 decrease the level of homocysteine, thus helping to control AD [23]. In a study of 156 mild cognitive impairment patients, Douaud and colleagues demonstrated attenuated gray matter atrophy using high doses of B vitamins over a period of 2 years [24]. The B vitamins appeared to lower the subjects’ homocysteine levels, resulting in reduced gray matter atrophy and slower cognitive decline [24]. However, a meta-analysis by Zhang and colleagues found that while intervention groups treated with vitamins B6, B12, and folate often experienced a reduction in homocysteine levels, these reductions were not translated into better cognition [25]. As demonstrated by these two studies, the effects of the B vitamins on cognition are uncertain. Thus, it is still unclear at this time whether vitamins B6, B12, or folate act as effective preventive or adjuvant treatments for AD.
Vitamin C
It is well established that individuals with mild cognitive impairment and AD have decreased plasma levels of ascorbic acid [26–29]. Vitamin C, or ascorbic acid, is theorized to hamper AD pathology and neurodegeneration by two main mechanisms: the modulation of oxidative stress and neuroinflammation [26, 30]. The anti-inflammatory properties of ascorbic acid are mainly attributed to its ability to block the lipopolysaccharide-stimulated production of inflammatory mediators [31], thus suppressing the MAPK and NF-κB systems [26, 32]. In addition, ascorbic acid supplementation reduces the amyloid plaque accumulation in AD mouse models [33–36]. An observational study reported that supplementation of antioxidant vitamins (C and E) was linked with decreased incidence and prevalence of AD [37].
Other beneficial actions include brain protection against glutamate-mediated oxidative stress or excitotoxicity [26, 38]. Excessive glutamate release leads to hyperactivation of N-methyl-d-aspartate (NMDA) receptors and neuronal damage. However, ascorbic acid prevents glutamate-mediated excitotoxicity by inhibiting the binding of glutamate to NMDA receptors [39–41].
Vitamin D
Vitamin D is linked to AD through regulation of calcium homeostasis, Aβ deposition, antioxidant, and anti-inflammatory properties [42–46]. In animal studies, researchers have demonstrated improved cognitive function and reduced amyloid burden in response to vitamin D supplementation [45, 47]. In a meta-analysis by Etgen et al. [48], the researchers proposed a nearly doubled risk of cognitive impairment for individuals with vitamin D deficiency. Furthermore, human genetic studies have demonstrated a clear relationship between overexpression of vitamin D receptors and reduced amyloid-β protein precursor expression [49, 50]. Vitamin D reduces the level of both Ca2 + and ROS by controlling the expression of Klotho/Nuclear factor (erythroid-derived 2)-like 2 (Nrf2), regulatory components of the calcium signaling system and redox signaling system. Reduction of Klotho/Nrf2 expression followed by a decline in vitamin D has been found in age-related cognitive decline in rats [51]. Miller and colleagues reported that vitamin D supplementation for the vitamin D insufficient adults increase plasma Aβ40, particularly in older adults, suggesting decreased brain Aβ [52]. The results from a large population-based study indicate that vitamin D intake in women with vitamin D deficiency show lower cognitive decline compared to the control group [53]. Furthermore, vitamin D as an adjuvant supplement to memantine treatment shows improved cognition among patients with AD more than memantine alone [54].
Vitamin E
Vitamin E exerts neuroprotective actions by modulating cognition through antioxidant and anti-inflammatory activity [49, 55]. A further study of the rat hippocampus found that vitamin E deficiency enhances Aβ deposition by reducing the expression of genes that code for proteins that indirectly or directly participate in Aβ clearance, such as insulin-degrading enzyme [56, 57]. For the first time, Santos and colleagues reported that vitamin E (2000 IU/d vitamin E or placebo for two years) slows AD progression [58]. Since then, several clinical studies have investigated the efficacy of vitamin E in AD treatment. However, some of the trials show no benefit [59–62].
Vitamin K
Evidence shows that individuals with AD often have low serum concentrations of vitamin K [63–65]. Furthermore, geriatric patients that use vitamin K antagonists as anticoagulant medications suffer more frequent cognitive impairment than patients not prescribed these medications [66]. Vitamin K is involved in neuron development and survival through anti-apoptotic and anti-inflammatory effects which are mediated by Gas 6, protein S, and sphingolipids [67]. Vitamin K may decrease AD risk by modulating sphingolipid metabolism, leading to enhanced Aβ clearance. Sphingolipids are a class of lipid molecules that confer specific characteristics to the membrane, thereby regulating subcellular trafficking and signaling pathways. Sphingolipids can promote the accumulation of Aβ in endosomal and lysosomal compartments [68]. Vitamin K deficiency can decrease the activity of the enzymes that are involved in sphingolipid metabolism, leading to improper sphingolipid metabolism, and ultimately, poor Aβ clearance and cognitive decline [56]. There is a lack of human clinical trials on the relationship between vitamin K and improvement in AD.
Despite a promising theoretical basis regarding the antioxidant and anti-inflammatory roles of vitamins, randomized clinical trials have not demonstrated a neuroprotective effect of vitamin supplementation on developing or preventing AD. Vitamin treatment could show an unreliable effect on AD because of differences in 1) the antioxidant and anti-inflammatory effect of vitamins in each person, 2) diet and nutritional status of the patients, 3) progression of the disease, and 4) types of vitamin deficiency of the patients. However, it has been determined that individuals with mild cognitive impairment and AD may have decreased plasma levels of one or more vitamins.
Random treatment of individuals with mild to moderate AD with vitamin C and E, α-lipoic acid, and coenzyme Q3 reduced oxidative stress in the brain; however, it did not influence cerebrospinal fluid biomarker related amyloid or tau pathology and also raised the potential issue of faster cognitive decline [69]. Cornelli reported that patients with AD treated with a cholinesterase inhibitor in combination with a low dose of antioxidants (carnosine, coenzyme Q10, vitamin E, vitamin C, beta-carotene, selenium, L-cysteine, vitamins B6, B9, and B12, and Ginkgo biloba) showed significant improvement [70]. The available evidence is insufficient and does not fully support the role of vitamins with cognitive decline regarding preventing or treatment of AD and further investigations are needed.
BALANCE OF OMEGA-3/OMEGA-6 FATTY ACIDS
The imbalance of omega-3/omega-6 fatty acids in the typical Western diet sparks harmful peripheral inflammatory processes [71, 72]. Evidence suggests that the actions of omega-3 promote an anti-inflammatory state, while the actions of omega-6 increase inflammation [71, 72]. Increased ingestion of omega-6 fatty acid leads to the overproduction of omega-6 derived signaling molecules (eicosanoids), which when produced in large amounts appear to contribute to the onset of an inflammatory state through a variety of mechanisms, including the release of pro-inflammatory cytokines [73]. Furthermore, increased omega-6 in the human diet coincides with the rising prevalence of obesity, a significant risk factor for AD development and progression that leads to widespread, systemic inflammation [71, 72]. A systematic review of 13 animal studies and 14 human studies supports the association between dietary omega-3/omega-6 fatty acids and the risk of developing AD [74]. The evidence supports the role of omega-3 as an anti-inflammatory fatty acid in preventing cognitive decline in AD at the early stage of the disease.
GLUTEN SENSITIVITY
Celiac disease (CD) is an autoimmune disease triggered by the ingestion of gluten that affects approximately 1% of the population [75, 76]. Recently, CD has come to be understood as only one of a possible range of clinical manifestations of gluten sensitivity diseases [75, 77]. Non-celiac gluten sensitivity (NCGS) is a syndrome characterized by symptoms related to the ingestion of gluten in the absence of CD or wheat allergy [77].
Non-celiac gluten sensitivity
NCGS may provoke AD pathology by initiating an overzealous immune system response following gluten ingestion which leads to a chronic state of inflammation [77, 78]. The first studies of NCGS indicated that only the innate immune system was involved in its pathogenesis [78, 79], but recent studies have detected the presence of anti-gliadin antibodies that may indicate the involvement of adaptive immunity [80, 81]. Diagnostic complications and ambiguous symptoms contribute to widespread disease mismanagement of NCGS, which prolongs the activation of the immune system and associated inflammation [81]. If it is accepted that NCGS promotes a chronic inflammatory state in a method analogous to obesity or traumatic brain injury [4], recognized risk factors for AD, then NCGS may lead to brain neuroinflammation and AD pathogenesis.
Celiac disease
In CD individuals, genetic susceptibility is present in the form of specific mutations to the major histocompatibility (MHC class II) alleles [82]. MHC class II molecules are present on antigen-presenting cells, where their role is to present antigenic peptides to other immune cells, like the T cells. Mutated forms of the gene can bind gluten peptides and activate T cells in the mucosa of the small intestine [82, 83]. Once activated, these T cells increase the production of the cytokine IFN-γ, which leads to mucosal damage of the small intestine [82, 85].
Individuals with CD display increased small intestine permeability as a consequence of autoimmune pathology [78]. The bacteria and fungi that colonize the digestive tract secrete amyloids, lipopolysaccharides [31], and other microbial exudates from their outer membranes [86–90]. Under conditions of increased intestinal permeability, these microbial amyloids and lipopolysaccharides may escape from the digestive tract and induce the immune system to increase the secretion of pro-inflammatory cytokines [88, 92]. Thus, increased intestinal permeability in CD may contribute to the onset of a chronic peripheral inflammatory state and thus the pathogenesis of AD. For further discussion of this topic, see the following section on the microbiome.
MICROBIOME
The microbiome influences the activity of the CNS, and the CNS influences the activity of the microbiome [93, 94]. Disturbances that alter the composition of the gut microbiome may stimulate various pathways that ultimately increase the risk of AD. Gut microbiota may be disturbed following exposure to antibiotic treatment, dietary changes, non-steroidal anti-inflammatory medications, food additives, various health conditions, and pathogenic infections [88, 95–99].
Increased gut permeability and bacterial secretions
Bacteria and fungi that colonize the human digestive tract secrete amyloids, lipopolysaccharides [31], and other microbial exudates from their outer membranes [86–90, 100]. For example, Escherichia coli produce extracellular amyloids, curli fibers that facilitate surface adhesion. Microbiome amyloid products such as CsgA, curli, and the Aβ42 peptides are recognized by TLR2/TLR1 receptors that are mediated pro-inflammatory responses [90]. Humans appear to sustain life-long exposure to large quantities of amyloid protein secreted by the gut microbiota, which may ultimately contribute to the pathogenesis of AD during aging [101–103]. Both aging and gut microbiota dysregulation contribute to an increase in the permeability or “leakiness” of the gut [94, 104]; thus, under certain conditions, these microbial amyloids and lipopolysaccharides may escape from the digestive tract and cause the immune system to increase secretion of proinflammatory cytokines [88, 92]. Increased proinflammatory cytokine secretion may contribute to the onset of insulin resistance, a recognized risk factor for AD [88, 105]. Additionally, an increase in the overall inflammatory state may itself contribute to the pathogenesis of AD.
Furthermore, the leaked bacterial-derived amyloids could cause an increase in ROS and subsequent activation of NF-κB, leading to upregulation of microRNA-34a. NF-κB is an essential mediator of inflammatory responses including expression of pro-inflammatory genes such as cytokines, activation, and differentiation of innate immune cells such as inflammatory T cells, and activation of inflammatory caspases such as NLRP3 inflammasome [106]. Furthermore, NF-κB increases the expression of microRNA-34, appearing to downregulate the expression of triggering receptors expressed on myeloid cells 2 (TREM2) [88]. Since TREM2 directly participates in the sensing and clearance of Aβ40 and Aβ42 peptides, it is probable that its downregulation will lead to impaired peptide phagocytosis and enhanced amyloid aggregation [88, 108].
In addition, alterations in the amounts of the neurotransmitters γ-aminobutyric acid (GABA), serotonin (5-hydroxytryptamine), brain-derived neurotrophic factor, and glutamate due to changes in microbiome status may contribute to the pathogenesis of AD [94, 109].
The hygiene hypothesis
The hygiene hypothesis posits that an overly hygienic Western lifestyle that includes the use of food additives, the abuse of antibiotics, clean drinking water, and a generally high level of sanitation, results in lower levels of infection and ultimately immune system dysfunction [94]. A new interpretation of the hygiene hypothesis, the microflora hypothesis, suggests that high sanitation alters the colonization of the infant gut, which disrupts the development of the immune system and leads to diseases [110]. While the mechanism of microbial modulation of host immunity is incompletely understood, some evidence suggests the role of T cells, specifically regulatory T cells, regarding immune response [94, 111–114]. Regulatory T cells act to suppress possibly harmful activities of the helper T cells, regulate the strength of the immune response [115–117], and protect commensal bacteria from elimination by the immune system [116, 118]. Inadequate exposure to microorganisms may fail to induce the differentiation of naïve T cells to regulatory T cells, rather than helper T cells [114]. Dysfunction of regulatory T cells has been suggested to partially modulate the relationship between decreased microbial exposure and the increased prevalence of allergic disease, autoimmune disease, and chronic inflammatory diseases in the rich, developed countries [94, 112]. Considering the parallels that can be drawn between AD and both the chronic inflammatory diseases and the autoimmune diseases, it is possible that the hygiene/microflora hypothesis may be applied as a framework to understand the rapid increase of AD pathology in recent years.
DISCUSSION
Genetics and unhealthy lifestyle habits, such as a noxious diet, can increase susceptibility to chronic inflammatory diseases [119, 120]. A Western diet induces systemic inflammation and alters innate immunity by reprogramming of myeloid progenitor cells to enhance immune responses [121]. However, one study suggests that the Mediterranean diet reduces the risk of AD and cognitive decline in individuals [122]. Scarmeas [123] also suggested that the Mediterranean diet reduces the risk of conversion from mild cognitive impairment to AD [124]. The beneficial components of the Mediterranean diet such as higher intake of fish, fruits, and vegetables rich in omega-3, olive oils, vitamins, phenols, and polyphenols provide antioxidant and anti-inflammatory properties, which are both important in the pathogenesis of AD [125–129]. The clinical trial, Three-City Study, also indicates that the Mediterranean diet is associated with a slower cognitive decline on the Mini-Mental State Examination. However, its association with the risk of dementia is not indicated [123]. Freat indicates that the major difference between the Three-City Study and Scarmeas’s study is the use of multi-vitamins and supplements, and also the length of follow-up regarding the risk of dementia. Furthermore, identification of the disease at the earlier stage of AD, by using new imaging technology, might provide a window of opportunity to use the protective effects of the Mediterranean diet against cognitive decline. By the time of clinical diagnosis of dementia and AD, it is likely too late to reverse the physiopathology of the disease.
The Mediterranean diet is a low carb diet with lean amounts of protein. Kroemer and colleagues characterize the excessive intake of carbohydrates that mediated noxious effects on human health as “carbotoxicity”. Low carb diets with adequate protein consumption cause ketogenesis, which is the conversion of body fat into ketones. Ketogenesis confers neuroprotective properties to the body through reduction of inflammation and oxidative stress, and increase in autophagy [130].
There is little known about the connection between gluten sensitivity and cognitive decline. However, nutrient deficiency, inflammation, and strong immune response in the brain puts patients with CD at higher risk of cognitive decline.
As the literature indicates, inflammation contributes to AD pathogenesis. Inflammatory pathways can accelerate the progression of AD [131] and several other age-related diseases [132]. An anti-inflammatory therapy should be beneficial in delaying the progression of AD. However, the results are far from conclusive [131]. It is important to know chronic inflammation is linked to the neurobiology and progression of AD [133] through different mechanisms, such as production and clearance of Aβ. Aβ plaques can trigger inflammation in a positive feedback loop [132].
CONCLUSION
Diet plays a significant role in introducing antioxidant and anti-inflammatory factors, shaping microbiomes, and treating digestive problems such as gluten sensitivity, that might decelerate inflammation and AD progression (Fig. 1). The inflammatory events throughout an individual's lifetime suggest the presence of a chronic inflammatory condition. The cumulative effects of this chronic inflammation may impact the different stages of AD [132]. Developing anti-inflammatory approaches to lifestyle habits may likely slow the progression, or delay the onset of AD. Health behavior interventions, such as dietary intake, may delay or slow the progression of AD [134–136] by 3 to 5 years [3]. Educating the public on delaying AD by early detection of the disease in combination with early intervention can act as effective methods to attenuate AD. Furthermore, interventional studies are necessary for considering whether a low antioxidant and anti-inflammatory diet contributes to the progression of AD and whether a high antioxidant and anti-inflammatory diet can reduce the risk of cognitive impairment and AD.
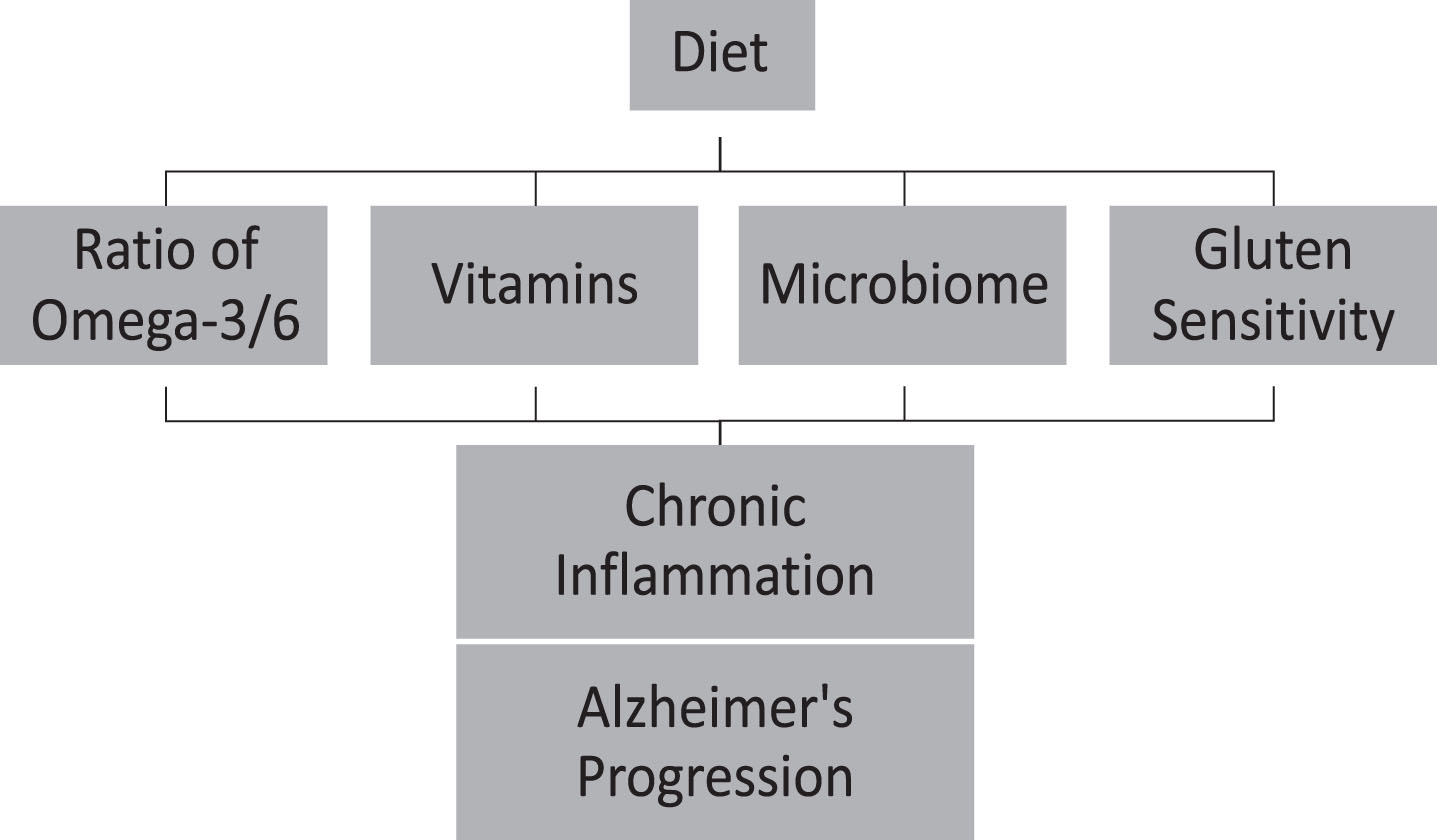
Diet can accelerate Alzheimer’s disease progression throughout chronic inflammation.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
Footnotes
ACKNOWLEDGMENTS
We would like to thank Dr. Stinson, Lamar School of Nursing Chair, and Dr. David Cocke, Gill Professor at Lamar University, for their support on this project.