
Abstract
Sensorimotor deficits have been described in several neuropsychiatric disorders including Alzheimer’s disease. The aim of the present study was to evaluate possible sensorimotor gating deficits in the Tg4-42 mouse model of Alzheimer’s disease using the prepulse inhibition task (PPI). Previous studies indicated that the hippocampus is essentially involved in the regulation of PPI. We analyzed 7-month-old homozygous Tg4-42 mice as mice at this age display severe neuron loss especially in the CA1 region of the hippocampus. Our results revealed a reduced startle response and PPI in Tg4-42 mice. The observed deficits in startle response and PPI are likely due to altered sensory processing abilities rather than hearing deficits as Tg4-42 displayed intact hearing in the fear conditioning task. The present study demonstrates for the first time that sensorimotor gating is impaired in Tg4-42 mice. Analyzing startle response as well as the PPI may offer valuable measurements to assess the efficacy of therapeutic strategies in the future in this Alzheimer’s disease model.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by cognitive and memory deficits due to structural and functional abnormalities including amyloid-β (Aβ) accumulation, neurofibrillary tangles and neuron loss [1–3].
Attention and executive function deficits as well as deficits in information processing are early characteristics of AD [4]. AD patients show a reduced attention span and are more prone to distractions due to impaired inhibitory mechanisms [5, 6]. The lack of filtering out unnecessary, conflicting or redundant information, a process known as sensorimotor gating, is an aspect of selective attention that is affected early in the disease progression [7, 8].
Prepulse inhibition (PPI) is a test considered to mirror an organism’s ability to filter out irrelevant information is used to assess attention deficits and sensorimotor gating. PPI refers to a reduction of the magnitude of the startle response when a stimulus (pulse) is preceded by a weaker stimulus (prepulse) [9, 10]. While PPI does not require learning, it is modulated by higher cognitive processes and PPI deficits reflect sensorimotor gating impairments and the inability to filter out irrelevant information [9, 11, 12]. Sensory gating deficits have been described in patients with mild and moderate AD and PPI has been discussed as a biomarker for early AD [13–15].
Acoustic startle response (ASR) is defined as a motor reaction to a loud and sudden acoustic signal and often measured in combination with PPI.
According to the amyloid cascade hypothesis, Aβ depositions are the causative event in the pathology of AD. Aβ is derived from the amyloid-β protein precursor (AβPP) through enzymatic cleavage by β- and γ-secretases [16]. Next to full-length Aβ starting with an aspartate at the first amino acid, a variety of truncated and modified Aβ species have been identified in AD. N-terminal truncated Aβ peptides are very abundant in the brains of familial and sporadic AD patients and there is substantial evidence that N-terminal truncated Aβ variants play a key role in the disease. Furthermore, N-terminal deletion enhances Aβ aggregation and neurotoxicity [17–19]. Thereby Aβ4-42 is a particular abundant species and one of the major fractions identified in the hippocampus and cortex of AD patients [20, 21]. The transgenic mouse model Tg4-42 expresses exclusively human N-terminally truncated Aβ4-42 albeit without human AβPP overexpression [22]. Tg4-42 mice show intraneuronal Aβ accumulation accompanied with astro- and microgliosis predominantly in the hippocampus [22]. In addition, Tg4-42 mice develop age-dependent behavior and memory deficits albeit without plaque formation [22, 23]. Furthermore, Tg4-42 mice display age-dependent motor deficits as well as an altered glucose metabolism accompanied with severe neuron loss [22–24].
In addition to Aβ plaques and neurofibrillary tangles, neuron loss is a main pathological hallmark of AD and cortical atrophy the most evident macroscopic characteristic of AD. [3]. Atrophy affects next to the amygdale and entorhinal cortex mainly the hippocampus [25, 26]. While the hippocampus is essential for the formation of memory and learning it has also been linked to sensorimotor processes. PPI, as a test for sensorimotor gating, is highly affected by the hippocampus [27]. It has been shown in different animal studies that alterations of the hippocampus lead to an impaired PPI [27–29]. Furthermore, hippocampal lesion and microinfusion studies demonstrated that the hippocampus plays a crucial role in regulating startle reactivity and PPI [29].
The aim of the current study was to extend previous findings on the Tg4-42 model by examining sensorimotor gating and acoustic startle response in aged Tg4-42 mice. Furthermore, we discuss the results in the context of other, well-studied AD models.
MATERIAL AND METHODS
Tg4-42 transgenic mice
The generation of Tg4-42 mice has been described previously [22]. Briefly, Tg4-42 mice express human Aβ4-42 fused to the murine thyrotropin-releasing hormone signal peptide under the control of the neuronal Thy-1 promoter. Tg4-42 mice are kept on a C57Bl/6J genetic background. Homozygous 7-month-old (±10 days) Tg4-42 and wildtype (WT) control mice (C57Bl/6J, Jackson Laboratories, Bar Harbor, ME, USA) were used in this study with an equal distribution of male and female animals (n = 11–13). No sex-differences were detected in any of the experiments. Mice used for the PPI study were part of a longer study and got injected intraperitoneal with a solution of 5% Tween80 in 0,9% sodium chloride daily for 6 weeks, starting at 3 months. Tween-80 is commonly used as a vehicle to evaluate the behavioral effects of experimental drugs without apparent adverse side effects [30–38]. All animals were handled according to the guidelines of the ‘Society for Laboratory Animals Science’ (GV-SOLAS) and the guidelines of the ‘Federation of European Laboratory Animal Science Association’ (FELASA). Furthermore, all experiments were approved by the ‘Lower Saxony State Office for Consumer Protection and Food Safety’ (LAVES). All efforts were made to minimize suffering and the number of animals used for this study.
Acoustic startle response and prepulse inhibition
Tg4-42 (n = 12) and WT (n = 13) animals were placed individually in a metal grid cage (90 x 40 x 40 mm) to restrict exploratory behavior and major movements. The cage was equipped with a movable platform floor attached to a sensor recording vertical movements (Process Control 25200 series, TSE GmbH, Germany). The cage was placed in a sound-attenuating isolation cabinet (TSE GmbH, Germany). Each experimental session started with a 3-min habituation period to 65 dB background white noise followed by a 2-min baseline recording. Loudspeakers were used to induce startle reflexes by acoustic stimuli. A startle reaction to an acoustic stimulus including body muscle contractions and jumping causes movement of the platform. A transient force resulting from this movement was recorded during a time window of 100 ms beginning with the onset of the acoustic stimulus.
Six pulse-alone trials using startle stimuli of 120 dB and 40 ms were applied after the baseline recording. PPI was tested applying the 120 dB 40 ms startle pulse alone or preceded by a prepulse 20 ms stimulus of 70, 75, or 80 dB. An interval of 100 ms with background noise was applied between each prepulse and pulse stimulus. A total of 30 trials with 10 trials of startle response alone, no stimulus trials and pulse preceded by a 70, 75, or 80 dB (2000 Hz frequency) prepulse were applied in a pseudorandom order with inter-trial intervals from 8 to 22 s. Maximum amplitudes for all types of trials were averaged for every mouse. Startle Response/PPI software Version 03.05 was used (TSE GmbH, Germany) and PPI at each sound level was calculated using the following formula:
Contextual and tone fear conditioning
An additional naive group of 7-month-old Tg4-42 (n = 12) and same-aged WT animals (n = 10) were subjected to a 3-day delay fear conditioning protocol as previously described [17]. Briefly, on the first day mice were allowed to explore a condition chamber (Ugo Basile, Italy) with black and white checkered walls and a steel grid for 150 s. Baseline freezing was measured during this habituation period using ANY-Maze video tracking software (Stoelting Co, USA). A tone (2000 Hz, 80 dB) was presented for 30 s that ended simultaneously with a 2-s foot-shock (0.7 mA). In order to test contextual memory mice were placed 24 h later in the same chamber for 210 s while freezing behavior was recorded. To test for tone fear conditioning 48 h later mice were placed back into the chamber now covered with white walls. Furthermore, an acetic acid scent was used to clean the chamber to create a new scent. After 150 s of baseline recording the same tone as in the fear conditioning trial was presented for 30 s without a following foot-shock. Freezing behavior was recorded before and during the tone.
Quantification of neuron numbers using unbiased stereology
Unbiased stereology was used to quantify the number of neurons in the hippocampal cell layer CA1 of 7-month-old mice (n = 10 per genotype) as previously described [22]. Briefly, mice were anesthetized and transcardially perfused with 4% paraformaldehyde. Left brain hemispheres were fixed in 4% paraformaldehyde, cryoprotected in 30% sucrose, frozen and frontally cut into a series of 30-μm thick sections on a cryostat (Microm HM550, Germany). Every tenth section was systematically sampled and stained with cresyl violet. Stereological analysis of the hippocampal cell layer CA1 (Bregma –1.22 to –3.80 mm) was performed Tg4–42 and WT mice using a stereology workstation (Olympus BX51 with a motorized specimen stage for automatic sampling, StereoInvestigator 7 [MicroBrightField, Williston, VT, USA]). The volume of the CA1 region was estimated by using Cavalieri’s principle [39].
Statistical analysis
Differences between groups were tested with unpaired t-test, one-way analysis of variance (ANOVA) or two-way analysis of variance (ANOVA) followed by Bonferroni multiple comparisons as indicated. Possible outliers were determined using the GraphPad outlier function. All data are given as means±standard error of the mean (SEM). Significance levels are given as follows: ***p < 0.001; **p < 0.01; *p < 0.05. All statistics were calculated using GraphPad Prism version 6.07 for Windows (GraphPad, USA).
RESULTS
Decreased acoustic startle response in Tg4-42 mice
The acoustic startle response (ASR) to the startle stimulus alone was measured in Tg4-42 and WT animals. Tg4-42 mice showed a significantly reduced startle response compared to same-aged WT mice (Fig. 1A; unpaired t-test: F(12,10)=8.148, p < 0.01). Furthermore, the latency to startle was significantly increased in Tg4-42 animals (Fig. 1B; unpaired t-test: F(12,10)=1.425, p < 0.001).
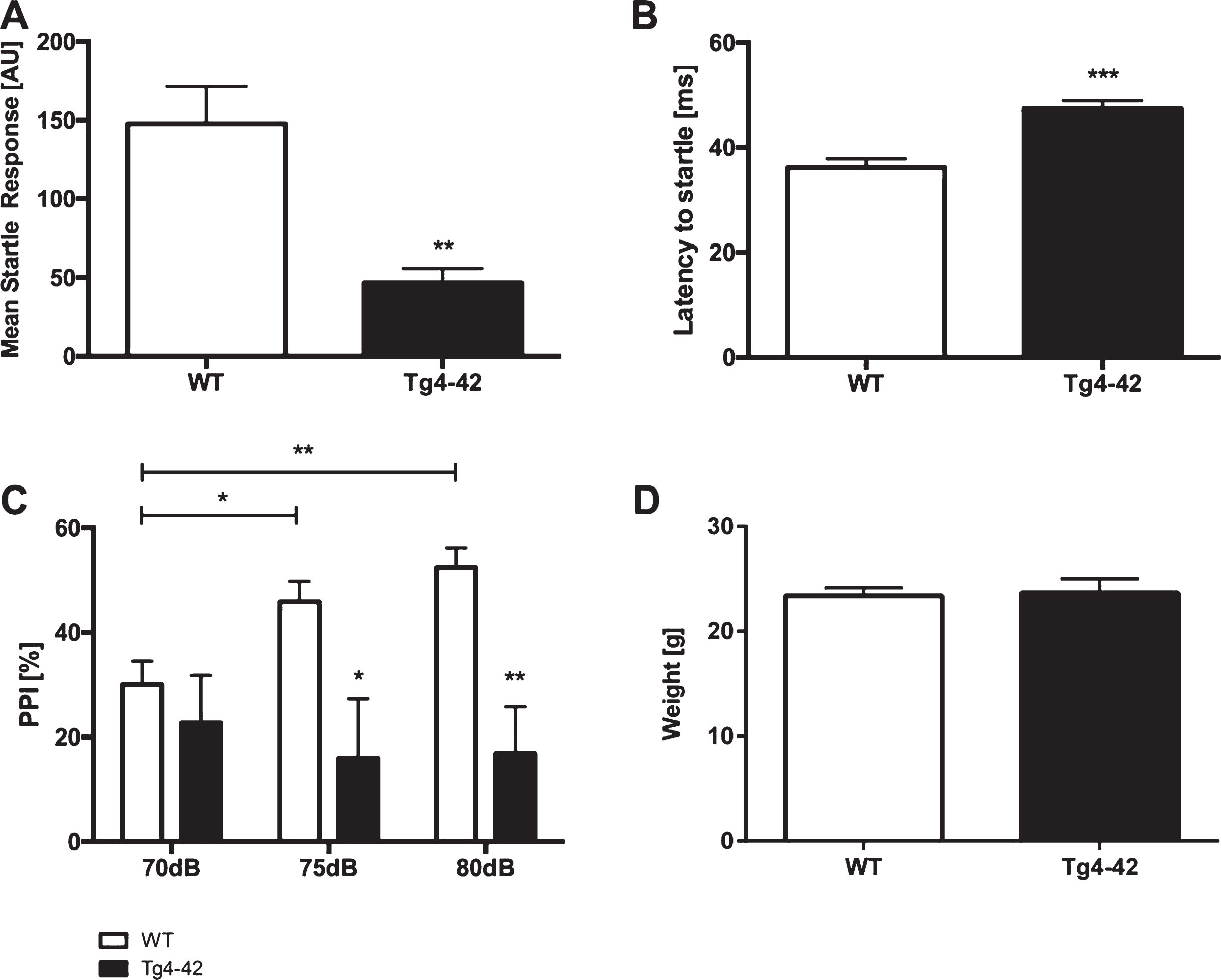
Altered acoustic startle response and prepulse inhibition in Tg4-42 mice. Tg4-42 mice showed a significantly lower acoustic startle response (A) and an increased latency to startle (B). Prepulse inhibition (PPI%) was significantly lower in Tg4-42 (n = 11) compared to WT (n = 13) mice at 70, 75, and 80 dB (C). Body weight did not differ between WT and Tg4-42 mice (D). AU = Arbitrary unit. Data presented as mean±S.E.M. A-B, D) unpaired t-test. C) Two-way analysis of variance (ANOVA) followed by Bonferroni multiple comparisons. ***p < 0.001; **p < 0.01; *p < 0.05.
Impaired prepulse inhibition in Tg4-42 mice
PPI was performed to analyze sensorimotor gating. Tg4-42 mice displayed a significantly lower PPI compared to same-aged WT animals at 70, 75, and 80 dB (Fig. 1 C; two-way-ANOVA, main effect of genotype: F(1,22)=12.05, p < 0.01). Louder prepulse tones did increase PPI in WT but not in Tg4-42 animals (Fig. 1 C; two-way-ANOVA, main effect of stimulus intensity: WT: F(2,36)=8.017, p < 0.01; Tg4-42: F(2, 30)=0.1378, p > 0.05). In addition, the weight of WT and Tg4-42 mice did not differ (Fig. 1D, unpaired t-test: F(15,13)=2.443, p > 0.05).
Decreased contextual learning in Tg4-42 mice
WT and Tg4-42 mice showed similar degrees of freezing in the training session of the fear conditioning experiment (Fig. 2A; one-way-ANOVA, training trial: F(3,42)=8.267, p > 0.05). However, when tested for context fear conditioning only WT mice demonstrated significantly increased freezing behavior (Fig. 2A; one-way-ANOVA, WT: F(3,42)=8.267, p < 0.001; Tg4-42: F(3,42)=8.267, p > 0.05). Tg4-42 showed impaired contextual learning as they did not associate the foot-shock during the training session with the chamber. It has to be noted that Tg4-42 animals showed normal pain perception as they vocalized and jumped in response to the electric foot-shock similar to WT mice.
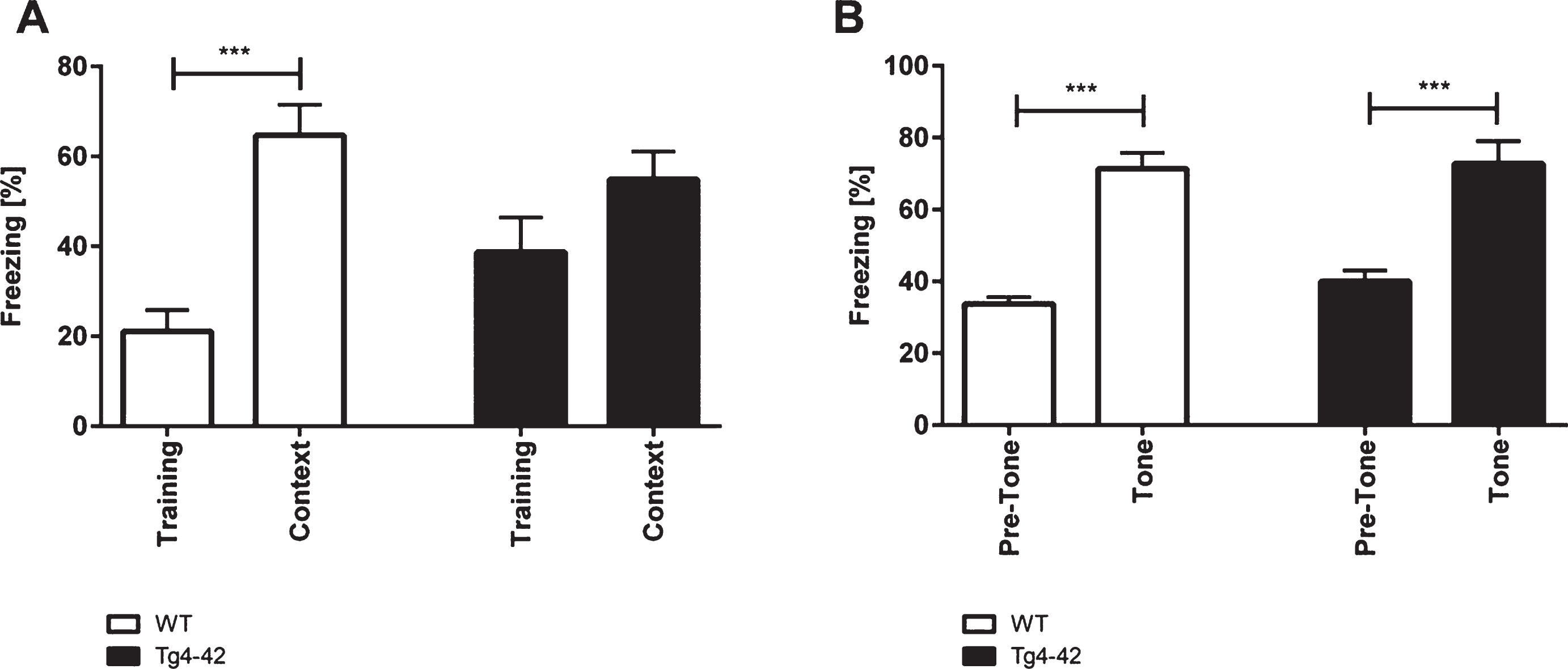
Impaired contextual conditioning in Tg4-42 mice. Tg4-42 and WT mice were trained in a contextual (A) and tone fear (B) conditioning task (n = 10-12). During the initial training session involving a tone-foot-shock pairing, WT and Tg4-42 mice displayed comparable degrees of freezing (A). Animals were reintroduced to the original training context 24 hours post training and tested for contextual memory. WT shock froze significantly more during re-exposure to the context compared to the training trial. In contrast, Tg4-42 mice did not associate the context with the received foot-shock as freezing was not significantly different between the training and the tone trial (A). Mice were placed in an altered fear conditioning chamber 48 hours post training and tested for freezing during tone presentation. WT and Tg4-42 mice showed a significant increase on freezing response to the tone presentation (B). Data presented as mean±S.E.M. One-way analysis of variance (ANOVA) followed by Bonferroni multiple comparisons. ***p < 0.001.
When tested for conditioned fear of a tone Tg4-42 and WT mice exhibited similar degrees of freezing as response to the tone (Fig. 2B; one-way-ANOVA, WT: F(3,42)=24.36, p < 0.001; Tg4-42: F(3,42)=24.36, p < 0.001).
Pronounced neuron loss in the hippocampus of Tg4-42 mice
Unbiased design-based Stereology revealed a 55% neuron loss in 7-month-old Tg4-42 mice in the CA1 layer of the hippocampus compared to same-aged WT animals (Fig. 3A; unpaired t-test: F(9,9)=1.343, p < 0.001; neuron number WT: mean = 286708±7670; neuron number Tg4-42: mean = 129713±6619). Furthermore, analysis of the CA1 volume demonstrated a significant volume reduction of 26% in Tg4-42 mice (Fig. 3B; unpaired t-test: F(9,9)=1.097, p < 0.05).
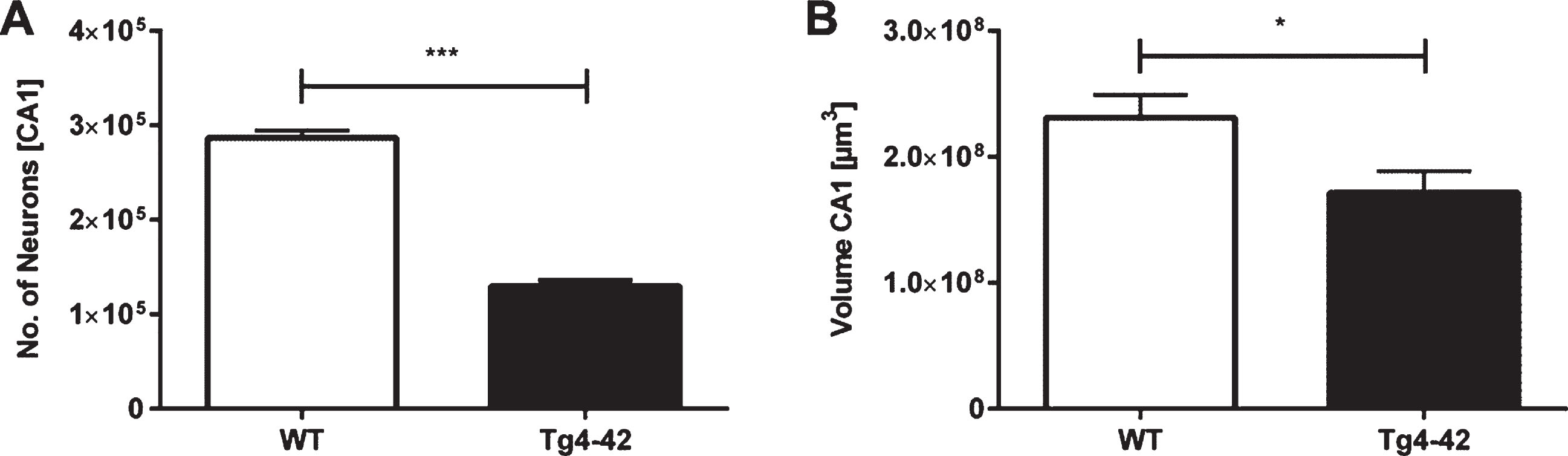
Neuron loss in the CA1 pyramidal cell layer of the hippocampus in Tg4-42 mice. Tg4-42 mice showed a significant neuron loss of 55% in the CA1 region (A). Furthermore, Tg4-42 mice displayed a 26% volume reduction in the CA1 region of the hippocampus (B). Data presented as mean±S.E.M. unpaired t-test. ***p < 0.001; *p < 0.05.
DISCUSSION
Acoustic startle response (ASR) is a protective response and defined as a motor reaction to a loud and sudden acoustic signal. While ASR in humans is mainly quantified using eye-blink reflexes, startle response in rodents is assessed through whole body reflexes [40–42]. Here we demonstrated that Tg4-42 mice showed a reduced startle response and an increased latency to startle. Interestingly, ASR seems to highly vary between different mouse models for AD. Mouse models of AD have shown unaltered, decreased as well as increased startle responses (Table 1). In line with our findings 5XFAD mice and Tau P301 S mice displayed reduced ASR [43–45]. In contrast, APOE knock-out and 3xTg mice showed an increased startle response [46, 47]. Several AD models including J20 and APP/PS1 also displayed an unaltered startle response [48–50].
Acoustic startle response and prepulse inhibition in different mouse models related to the Alzheimer’s disease pathology
ASR, acoustic startle response; PPI, prepulse inhibition; ns, not significant; NIA, no information available.
While ASR studies in AD patients are limited, Salem et al. (2001) [51] described an increased latency to peak startle response with increasing white matter hyperintensities that are highly associated with AD [52–54].
Analyzing ASR in Tg4-42 mice may offer a valuable measurement when assessing the efficacy of therapeutic strategies in this mouse model. Assessing startle response is a sensitive, non-invasive, reproducible, reliable and easy measure that can even be studied without analyzing PPI [13].
Sensorimotor gating can be measured using PPI, a method that can be studied with similar procedures in humans and rodents and reflects the ability to exclude sensory information from processing [55]. Reduced PPI reflects a dysfunction of sensorimotor gating and has been reported in several psychiatric and neurological disorders, including schizophrenia, post-traumatic stress, bipolar disorders, Huntington’s disease as well as AD [11, 56–59].
Interestingly, a large-scale meta-analysis from more than 1300 C57BL/6J male mice showed that there is no simple linear relationship between ASR and PPI levels [60]. In line with these findings, reduced PPI has been described in AD mice with an unaltered [48–50], reduced [43, 44], and increased startle response [47].
Tg4-42 mice demonstrated impairments of sensorimotor gating as PPI was significantly lower in transgenic mice compared to WT animals. Similar PPI deficits were reported in several AD lines including J20, TBA2.1, APP/PS1, and 3xTg AD mice (Table 1).
In line with our finding, Ueki et al. (2006) [14] reported sensorimotor gating deficits in patients with mild AD using PPI. In a recent study, PPI confirmed sensorimotor gating deficits in early AD cases [13]. However, another PPI study did not report differences between AD patients and healthy controls [61]. The inconsistent PPI results in AD patients are likely due to different study protocols and disease stages.
Previous studies indicate that the hippocampus is essentially involved in the regulation of PPI [27–29, 62]. Some of the earliest damages in AD brains have been reported in the hippocampus and Padurariu et al. (2012) reported a decrease in neuronal density particularly in the CA1 region of the hippocampus in AD patients [63]. Consistent with these results, Tg4-42 mice showed a neuron loss of more than 50% in the CA1 region of the hippocampus at 7 months of age. It has been demonstrated that hippocampal lesions induce disruptions of PPI in rodents [27, 64]. In line with our findings Caine and colleagues (1992) examined the relationship between PPI and the hippocampus and showed that modulations of the CA1 reduced the acoustic startle reflex and PPI in rats [28].
ASR and PPI can among others be affected by housing conditions, the genetic background strain, weight and hearing ability [65–69].
Weight can severely influence the outcome of behavior studies. Fodor et al. (2016) demonstrated that lower body weight in rats resulted in a reduced ASR, while body weight did not affect PPI [68]. Therefore, the animal’s weight should always be taken into consideration especially when analyzing startle response in rodents. However, the observed reduced ASR in Tg4-42 cannot be attributed to an altered body weight as the weight of transgenic mice was comparable to wildtype animals.
Willot et al. (2003) analyzed the correlation between acoustic startle response, PPI, and auditory abilities in forty inbred mouse strains and concluded that a severe hearing loss is necessary to influence PPI [65]. O’Leary et al. (2017) reported an age-dependent hearing dysfunction in 5XFAD mice and attributed the decreased startle response at least partially to the observed hearing loss [44]. In contrast, Story et al. (2018) argued that the decreased startle response observed in 5XFAD cannot be attributed to hearing deficits as 5XFAD mice show a response to the startle tone [43].
The altered startle response and impaired PPI in Tg4-42 mice might also be explained by possible hearing dysfunctions. If Tg4-42 mice display hearing impairments, then it could be argued that difficulties in detecting the prepulse could explain the impaired ASR and PPI. However, there is no evidence that Tg4-42 mice display any hearing deficits. Although Tg4-42 mice showed a decreased ASR they did respond to the 120 dB startle tone. In addition, Tg4-42 mice reacted to the 80 dB tone presented in the fear conditioning experiment similar to wildtype animals.
While Tg4-42 mice showed learning deficits in the contextual fear condition task, they displayed no impairments in conditioned learning in response to the tone stimulus. Homozygous Tg4-42 mice in this study exhibit a selective impairment of contextual fear learning while their tone learning ability remains intact similar to the results previously described in 12-month-old hemizygous Tg4-42 mice [70]. These results indicate that Tg4-42 mice exhibit a selective impairment of contextual fear learning while their tone learning ability remains intact. Therefore, the observed deficits in startle response and PPI are likely due to altered sensory processing abilities rather than hearing deficits. However, further studies are needed to assess hearing in Tg4-42 mice and determine the contribution of possible reduced auditory thresholds and non-auditory phenotypes on the acoustic startle response and impaired PPI.
The variability in experimental procedures between the tested AD models is compared to other behavior experiments rather small. The startle impulse used in different studies differed between 110 dB and 125 dB. The used prepulse tones ranged from 73 dB to 95 dB and the majority of studies used multiple prepulse tones with at least a 10 dB difference between the lowest and highest tone (Table 1). However, these varieties need to be taken into consideration when comparing different AD mouse lines.
Taken together, the present study demonstrates for the first time that sensorimotor gating is impaired in Tg4-42 mice at a disease stage where mice display significant neuron loss and memory deficits. Therefore, analysis of startle response and PPI offer a useful additional tool to measure therapeutic efficacy in Tg4-42 mice.
CONFLICT OF INTEREST
The Tg-4-42 mouse model has been patented by the University Medicine Göttingen and TAB.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the German Research Foundation (CNMPB) and the ‘Alzheimer Stiftung Göttingen’ to YB. This study was also supported by the Jacob-Henle-Program for Experimental Medicine of the University Medicine Goettingen to MJL. MES is supported by Helios Kliniken GmbH. We acknowledge support by the Open Access Publication Funds of the Göttingen University.