
Abstract
The posteromedial cortex (PMC) and medial temporal lobes (MTL) are two brain regions particularly vulnerable in Alzheimer’s disease (AD). We have reviewed the spatiotemporal patterns of amyloid-β and tau accumulation, local MTL functional alterations and MTL-PMC network reconfiguration, and propose a model to relate these elements to each other. Functional and structural MTL-PMC disconnection happen concomitant with amyloid-β plaques and neurofibrillary tau accumulation within these same regions. Ongoing disconnection is accompanied by dysfunctional intrinsic local MTL circuit hyperexcitability, which exacerbates across distinct clinical stages of AD. Our overarching model proposes a sequence of events relating the spatiotemporal patterns of amyloid-β and tau accumulation to MTL-PMC disconnection and local MTL hyperexcitability. We hypothesize that cortical PMC amyloid-β pathology induces long-range information processing deficits through functional and structural MTL-PMC dysconnectivity at early disease stages, which in turn drives local MTL circuit hyperexcitability. Intrinsic local MTL circuit hyperexcitability subsequently accelerates local age-related tau deposition, facilitating tau spread from the MTL to the PMC, eventually resulting in extensive structural degeneration of white and grey matter as the disease advances. We hope that the present model may inform future longitudinal studies needed to test the proposed sequence of events.
Keywords
INTRODUCTION
Background and rationale
Amyloid-β plaques and neurofibrillary tau deposits define Alzheimer’s disease (AD) as a unique disease among several that can lead to dementia [1]. AD is the most common dementia-causing neurodegenerative disease worldwide, characterized by progressive cognitive decline, often involving declarative memory deficits. In the classical and most frequently occurring amnestic form, pathological changes converge on the default mode network (DMN), a large-scale network associated with self-referential processes, including consolidation of episodic memory and future thinking (Fig. 1). The posteromedial cortex (PMC) is a key node of the DMN and composed by associative brain regions such as the precuneus, posterior cingulate, and retrosplenial cortices. These regions are implicated in a diverse range of higher-order cognitive functions including memory consolidation and spatial navigation [2]. The PMC is functionally and structurally connected to the medial temporal lobes (MTL), as revealed by studies of resting-state fMRI [2], diffusion tensor imaging studies tracking axonal tracts [2], and anatomical studies in the human and macaque brain [2]. The MTL comprises the hippocampal formation, the entorhinal cortex, the perirhinal cortex, and the parahippocampal cortex [3]. These subregions cohesively form a feedback circuit underlying memory processing through interplay with neocortical areas such as the retrosplenial and posterior cingulate cortices from the PMC [3]. The functional and structural connections between both regions are disrupted across distinct clinical stages of AD, and take place with simultaneous alterations in local MTL functional organization.
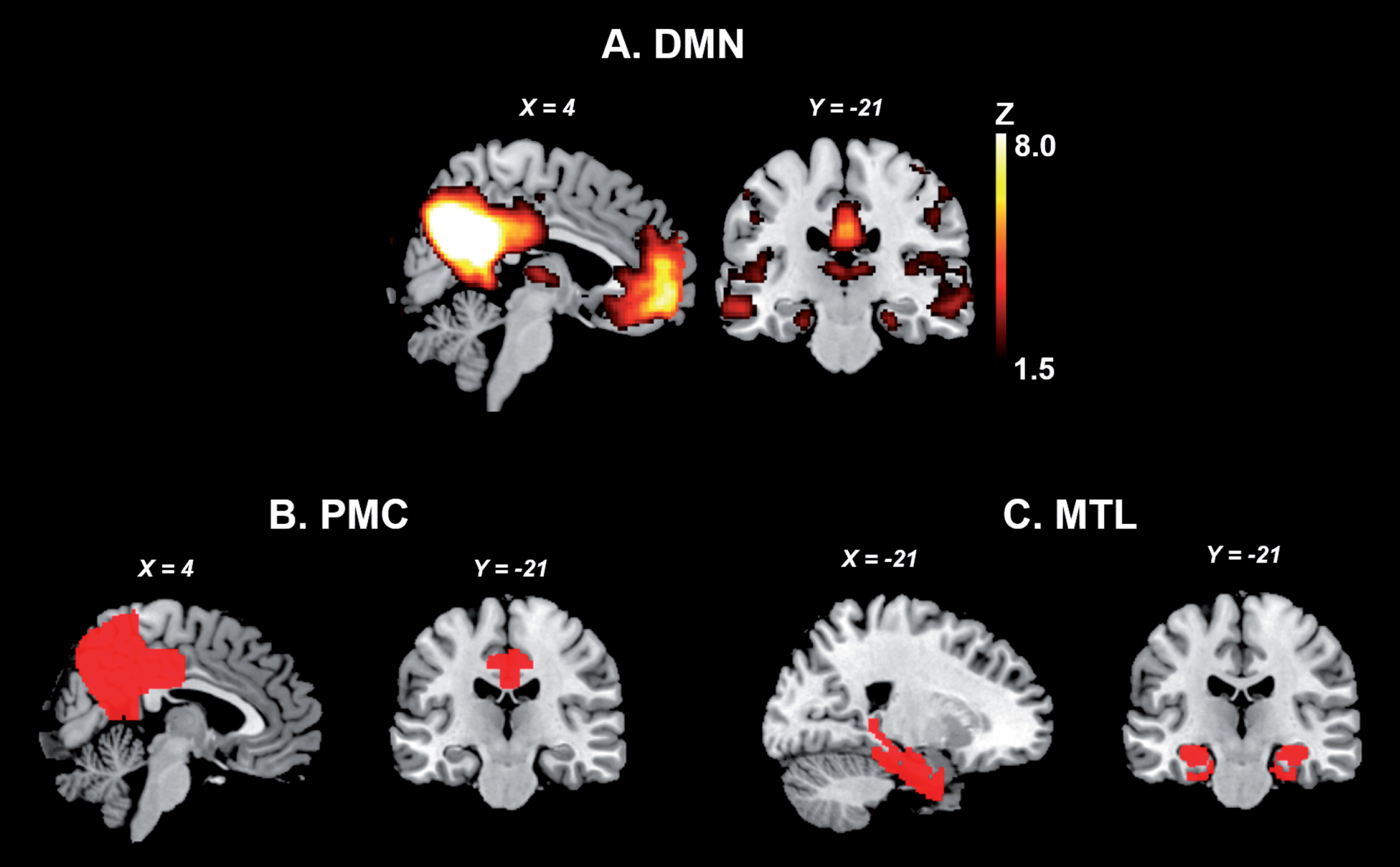
The MTL and PMC of the DMN. A. Publicly available spatial map of the DMN derived from an independent component analysis on resting-state fMRI data of 36 healthy subjects (https://www.fmrib.ox.ac.uk/datasets/brainmap+rsns/). B and C show the location of the PMC and MTL, respectively (Harvard-Oxford Cortical and Subcortical Atlas). DMN, default mode network; MTL, medial temporal lobes; PMC, parietomedial cortex.
The PMC and MTL are particularly vulnerable to AD-related pathological changes such as tau accumulation, amyloid-β deposition, hypometabolism, and grey matter atrophy, since in these areas early pathological changes converge across AD stages from preclinical AD, to mild cognitive impairment (MCI), to AD dementia [2, 4–6]. More precisely, on the one hand, tau has been shown to aggregate first in the locus coeruleus and then prominently in the transentorhinal cortex of the MTL, further spreading to limbic and isocortical areas [7]. On the other hand, amyloid-β deposition is prominent in the precuneus and posterior cingulate cortex, and is found in limbic and subcortical brain areas only at later disease stages [8]. Although the relationship between amyloid-β and tau deposition is poorly understood, current models propose that cortical amyloid-β deposition facilitates age-related tau deposition in the MTL, with tau subsequently spreading to neocortical regions following a network based pattern [9, 10].
The present review intends to: 1) propose a sequence of events linking MTL-PMC disconnection with local MTL dysfunction along the clinical trajectory of AD; and 2) propose a model where the distinct spatiotemporal patterns of tau and amyloid-β accumulation drive MTL-PMC disconnection and local MTL functional alterations. This effort aims to motivate future longitudinal studies reassessing the role of the MTL and PMC in AD pathogenesis, and to identify potential disease modifying interventions that target the mechanistic relationship between local MTL dysfunction and MTL-PMC disconnection.
Tracking down MTL-PMC disconnection
MTL-PMC disconnection in AD, which we define as an umbrella term encompassing early functional and structural alterations reflecting long-range deficits in information flow and late stage white matter degeneration, is concomitant with disease protein accumulation within these same regions. Measures of functional dysconnectivity (reduced correlated activity between distinct brain regions assessed with resting-state fMRI), as well as structural dysconnectivity (reduced integrity of axonal bundles assessed with diffusion tensor imaging), and white matter atrophy (see Box 1 for a summary of used terms) have been used to explore the integrity of the MTL-PMC system in AD. These measures consistently reveal MTL-PMC disconnection across clinical stages of AD [4, 11–18] (Fig. 2B), with functional changes preceding structural ones, possibly reflecting dysfunctional integration between both systems preceding neuronal death and atrophy (Fig. 2A) [4, 17–22]. Evidence of MTL-PMC disconnection is even found in subjects at higher risk of developing dementia such as apolipoprotein E4 (APOE4) carriers, amyloid-β positive, cognitively normal individuals (preclinical AD), and patients with amnestic MCI [23, 24], although contrasting findings have been published so far for cognitively healthy APOE4 carriers with other works reporting increased long-range connectivity in this population [25].
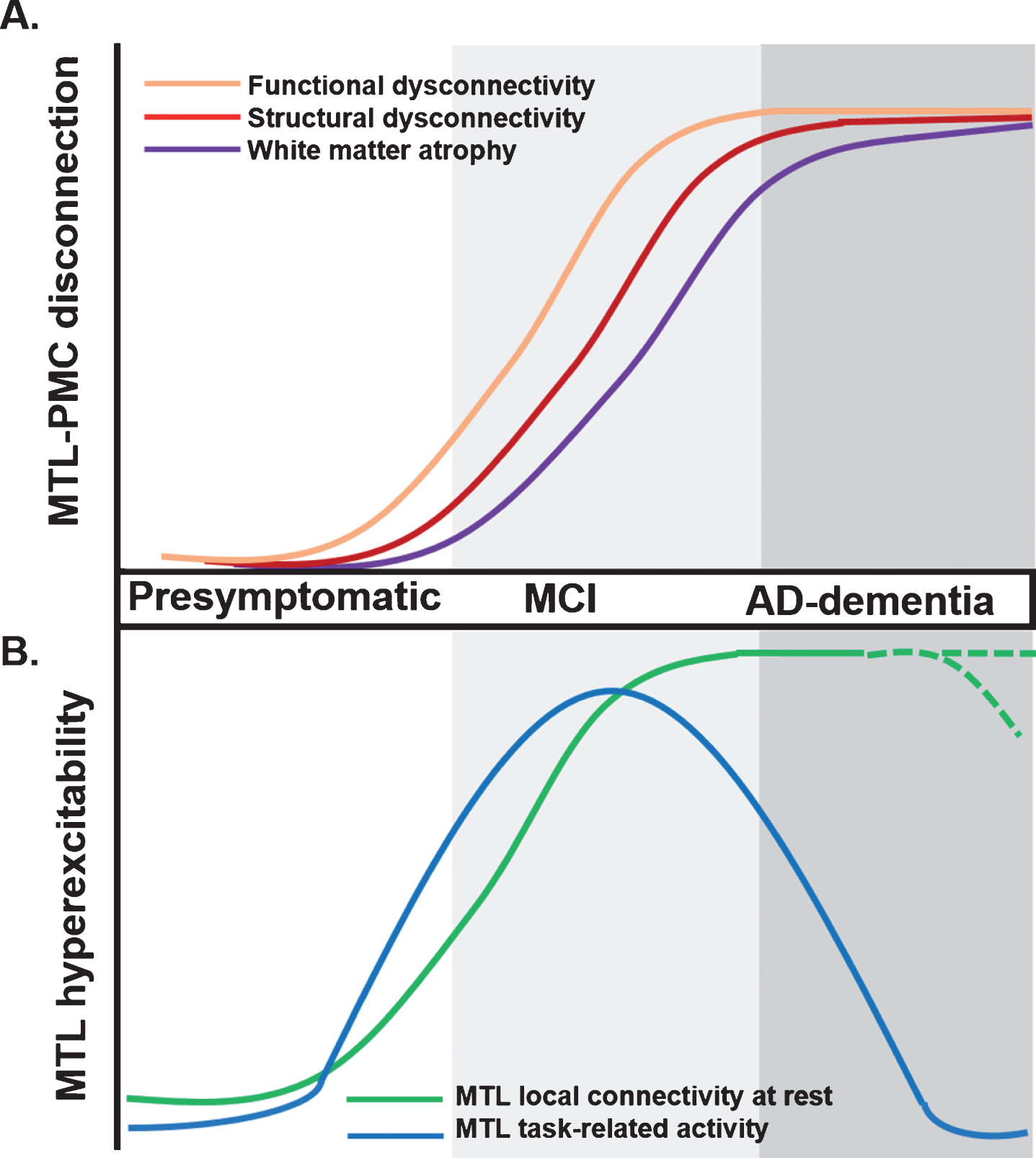
Proposed trajectory for MTL-PMC disconnection and MTL circuit hyperexcitability across clinical stages of AD. A) MTL-PMC disconnection progressively increases across the clinical AD trajectory (indicated by shades of grey), starting with functional (orange curve) and followed by structural dysconnectivity (red) reflecting dysfunctional communication between both regions but not yet extensive neuronal death and degeneration at early AD stages. Functional and structural dysconnectivity are eventually followed by disconnection measured through white matter atrophy reflecting overt degeneration (violet). B) In parallel to ongoing MTL-PMC disconnection, local MTL functional hyperexcitability takes place across stages of AD. On the one hand, task-related MTL activity follows an inverse-U-shaped activation trajectory, with hyperactivity patterns in early MCI followed by hypoactivity at later disease stages (blue hyperbola). On the other hand, during resting-state conditions the MTL is characterized by progressive local hyperconnectivity across clinical AD stages (green curve). It is currently unknown whether at final stages, MTL hyperconnectivity is sustained or eventually drops due to ongoing MTL degeneration (shaded green lines). AD, Alzheimer’s disease; MCI, mild cognitive impairment; MTL, medial temporal lobes; PMC, parietomedial cortex.
Meanwhile, studies focusing on the MTL suggest an inversed-U-shaped pattern of activation in the MTL during memory tasks across clinical AD stages [26, 27]. Although in healthy APOE4 carriers and MCI patients discrepant findings of both task-related MTL hypoactivation and hyperactivation have been reported [25, 28–33], MCI patients with lower dementia score as determined by Clinical Dementia Rating show paradoxically increased task-related hyperactivity in the MTL compared to healthy controls and advanced dementia [34]. In contrast to findings in early MCI, local MTL hypoactivity patterns are found during memory tasks at later disease stages. Importantly, low doses of the antiepileptic drug levetiracetam, normalize task-related hyperactivity in the hippocampus and improve memory performance in patients with MCI [27]. It is important to note that under resting-state conditions, increased local functional connectivity and increased magnitude of slow brain activity fluctuations are found within the MTL across clinical AD stages [11, 35]. Local MTL functional alterations correlate with MTL-PMC disconnection in functional and structural terms [12, 36]. The nature of these local MTL functional alterations is currently unclear, with some studies suggesting MTL functional reorganization to reflect compensatory mechanisms of the MTL in interaction with other brain areas in order to sustain compromised cognitive functions [33, 37]. Alternatively, based on 1) the normalizing effects of levetiracetam, 2) the link to MTL disconnection, and 3) the positive association with memory impairments, altered patterns of local MTL functional organization have been proposed to reflect disconnection-based dysfunctional imbalances in the MTL circuit that drive elevated levels of local activity and synchronicity, which we from now on will describe as MTL circuit hyperexcitability [11, 36].
OUR MODEL
We propose a sequence of events relating MTL-PMC disconnection and local MTL functional alterations in AD. This model has been inspired by previously published hypothetical models describing the temporal evolution of AD biomarkers in relation to each other and to onset and progression of clinical symptoms [38], which hypothesized that incipient amyloid-β pathology could accelerate antecedent tau pathology [4, 38–41]. We propose that cortical PMC amyloid-β pathology is the initial factor facilitating MTL-PMC structural and functional dysconnectivity at initial disease stages, reflecting dysfunctional long-range communication between both brain areas but not yet neuronal death and overt degeneration. MTL-PMC dysconnectivity inevitably drives local MTL network hyperexcitability, resulting in local hyperconnectivity patterns that in turn exacerbate local age-related tau deposition which has been taking place independently from amyloid-β pathology [42, 43]. We hypothesize that MTL network hyperexcitability is a critical factor accelerating tau pathology accumulation and spread from the MTL to the PMC, eventually resulting in extensive MTL-PMC disconnection reflected by white and grey matter atrophy (Fig. 3).

AD model linking MTL-PMC disconnection and MTL circuit hyperexcitability with tau and amyloid-β pathology. The sequence of events schematized in panels A and B are complementary and do not mutually exclude each other. A) At initial disease stages, amyloid-β pathology impairs the normal information flow between the MTL and PMC by driving functional and structural dysconnectivity between the PMC and the MTL, which results in disinhibition of the local MTL circuit and local MTL hyperactivity and hyperconnectivity during task and rest. MTL circuit hyperexcitability is the driving force accelerating age-related tau accumulation and atrophy in the MTL. B) Tau accumulation and atrophy in the MTL increase levels of local MTL circuit hyperexcitability, resulting in additional tau accumulation and facilitating subsequent tau spread out from the MTL to the PMC. Tau spread from the MTL to the PMC leads to advanced MTL-PMC disconnection through degeneration of white-matter tracts, eventually followed by cortical atrophy of the PMC. MTL, medial temporal lobes; PMC, parietomedial cortex.
Early experimental and observational data
Task- and resting-state related functional alterations in the MTL have been repeatedly associated with MTL-PMC disconnection [11, 12], atrophy [26, 36], memory dysfunction [11, 27], and amyloid-β pathology [44], suggesting dysfunctional rather than compensatory functional changes in AD. We propose that at initial stages of the disease (Fig. 3A), amyloid-β pathology in the PMC facilitates functional and structural dysconnectivity between the PMC and the MTL reflecting dysfunctional interactions between both brain regions but not yet overt neurodegeneration [6]. Dysconnectivity inevitably results in disinhibition of the local MTL circuit and respectively in local MTL hyperactivity and hyperconnectivity during task and rest [11, 44]. Deleterious effects of local MTL age-related tau deposition are facilitated by MTL circuit hyperexcitability. Local MTL circuit imbalances further contribute to MTL-PMC dysconnectivity, which in a vicious cycle exacerbate the observed patterns of early stage MTL circuit hyperexcitability at rest [11, 43]. Such levels of intrinsic MTL circuit hyperexcitability may add to the relatively preserved task-related activity of the MTL in early AD, yielding to MTL hyperactivity patterns observed during memory tasks in MCI [27].
Local circuit hyperexcitability of the MTL could be the driving force accelerating amyloid-β independent age-related tau accumulation in the MTL [42, 43] (Fig. 3B), leading to further MTL disinhibition, and subsequent tau spread from the MTL to the PMC. The synergistic action of MTL-PMC dysconnectivity, MTL circuit hyperexcitability and additional tau accumulation, drives excitotoxic and neurodegenerative processes leading to atrophy of the MTL [42, 43]. Eventually tau spread from the MTL to the PMC is followed by widespread disconnection in form of degeneration of white-matter tracts connecting the MTL and the PMC [18, 45], culminating in cortical atrophy of the PMC [5, 26]. Importantly, with deteriorating disconnection at more advanced disease stages due to progressive tau accumulation and spread, activity in the MTL is progressively disinhibited, while the modulatory input from the PMC needed to sustain normal memory function is severely curtailed. This mechanism could explain the progressive increases in local MTL hyperconnectivity, beginning in MCI stage, while task-related MTL activity declines in more advanced disease stages.
Future experiments and validation studies
The model we propose here has important implications. The combination of multi-center longitudinal studies following subjects at different clinical stages of AD, together with multimodal neuroimaging tracking MTL-PMC disconnection, MTL circuit hyperexcitability, amyloid-β pathology, and most recently tau pathology, make some of the proposed assumptions in the model testable. Of particular interest are markers of tau pathology, with recent PET studies showing that tau accumulation predicts cortical atrophy in the entorhinal cortex, middle temporal gyrus and parahippocampal gyrus of the MTL, as well as in the precuneus and posterior cingulate gyrus of the PMC [46]. In line with our model and proposed experimental framework, Jacobs and colleagues have recently used longitudinally acquired neuroimaging data in amyloid-β positive healthy participants to show that microstructural alterations of the hippocampal part of the cingulum predicted PMC tau accumulation [47]. Alternatively, CSF markers of amyloid-β and tau pathology could be used to validate our model, since previous studies have demonstrated an association between CSF tau levels and; 1) decreased MTL microstructural connectivity, in healthy subjects at risk for AD [45], 2) lower connectivity of the cingulum fiber in patients with MCI [48], and 3) default mode network white matter degeneration in AD patients. In particular, available longitudinal datasets could help elucidate the temporal relationship between distinct components of the model, and could contribute to assess whether amyloid-β and tau pathology are necessary or sufficient conditions for MTL circuit imbalances and MTL-PMC disconnection. Our model highlights the critical role of MTL circuit hyperexcitability, not only as a result of AD neuropathology, but as a factor contributing to AD pathology spread to neocortical regions and ultimately neurodegeneration. This emphasizes the role of tau in induction and potentiation of MTL circuit hyperexcitability, leading to consequent atrophy in AD signature areas.
OUR MODEL IN THE CONTEXT OF LITERATURE
We propose that two main complementary mechanisms could mediate the deleterious effects of tau and amyloid-β, on progressive MTL-PMC disconnection and local MTL circuit hyperexcitability in AD [49]. In the first mechanism, pathology spreads form onset regions following a network-based pattern [50], resulting in neurodegenerative processes eventually leading to degradation of brain networks and the underlying functional and structural architecture [51]. Such “trans-neuronal” spread leads to direct neuronal dysfunction and results in progressive degrees of functional and structural disconnection. In AD, tau pathology seems to be a fitting candidate for such a mechanism, given that studies in rodents and humans provide evidence that tau spreads out from the entorhinal cortex to the rest of the brain in a “prion-like” fashion following brain network boundaries [52]. A second “wear-and-tear” [53] mechanism proposes that locally accumulating pathology results in noisy and inefficient synaptic communication in vulnerable brain regions with high metabolic demand, which shifts to downstream areas within highly-connected regions, leading to cascading network failure and disconnection [49, 54]. Evidence for such mechanisms comes from rodent models of AD, where local amyloid-β has been recently shown to elicit hippocampal hyperactivity and disrupt long-range hippocampal-cortical functional connectivity [55, 56]. Further works in transgenic mice models have provided evidence for disrupted cross-laminar cortical processing in presence of amyloid-β pathology [40], and have revealed that terminal fields in local and distant cortical areas contain numerous swollen dystrophic neurites often grouped in grape-like clusters in proximity of plaques [41]. In line with these findings, recent neuroimaging studies reported a negative correlation between local levels of amyloid-β and locally confined functional connectivity of several heteromodal intrinsic brain networks. This negative correspondence was found across clinical stages of AD, and was particularly evident within DMN nodes with high metabolic demand, such as the PMC [36]. Furthermore, tau has also been found to impair synaptic function, with extracellular tau impairing long-term potentiation and memory in mice [57]. Antisense reductions of tau in mice have been shown to protect against seizures [58], and tau pathology has been associated with severity of seizures in patients with status epilepticus and found postmortem in patients with temporal lobe epilepsy [59–62], providing additional evidence that tau may impair the excitatory-inhibitory balance in the MTL. A recent neuroimaging study combining tau-PET and resting-state fMRI in AD patients, has provided evidence for an association between tau pathology in the entorhinal cortex of the MTL and cascading network failure, expressed in terms of intrinsic functional dysconnectivity of several DMN subsystems [63]. Recently, neuroimaging studies have proposed a stage-dependent connectome mediation framework, where early local pathological changes impair neuronal function, followed by connectome-mediated effects on remote areas at later disease stages [64]. Our model unifies this framework with theories of AD by proposing putative mechanisms accounting for amyloid-β facilitation of MTL tau pathology and spread. In conclusion, our model provides a unitary framework in which neuronal mechanisms based on large-scale brain network reconfiguration and local MTL dysfunction mediate the link between cortical amyloid-β deposition and age-related MTL tau accumulation and spread [9].
CONCLUSIONS
Our model proposes a sequence of events linking the spatiotemporal accumulation pattern of AD disease proteins with selective vulnerability of the MTL and PMC. Beside the mechanistic value of the model, tracking the neuroimaging-based measures of MTL circuit hyperexcitability and MTL-PMC disconnection could be helpful for monitoring disease progression and interventions targeting MTL circuit hyperexcitability. Interestingly, similar patterns of MTL circuit hyperexcitability during rest have been found in patients with medial temporal lobe epilepsy [65]. This insight, together with the normalizing effect of antiepileptic drugs on MTL activity and memory performance in MCI, implicates that timely identification and treatment of MTL circuit hyperexcitability in MCI patients might prevent progression to AD dementia. The current framework, however, has several challenges. For instance, our model is based mostly on currently available cross-sectional data, urging for longitudinal studies corroborating or disproving the proposed sequence of events. In particular, our sequence of events is inspired by models proposing that amyloid-β pathology is influencing and accelerating tau accumulation and spread [9, 10]. However, it is important to note that alternatively tau pathology could be the initial factor actually leading to MTL-PMC disconnection and consequently PMC amyloid-β accumulation. More longitudinal data is needed to support either model. Second, our model is centered on the MTL and PMC, and ignores hence all other regions that are vulnerable to AD pathology, such as brainstem regions and medial prefrontal areas [7]. Most importantly, our model needs to be adapted to other clinical phenotypes, such as non-amnestic manifestations of AD, including posterior cortical atrophy and logopenic primary progressive aphasias [66]. However, recent neuroimaging studies have revealed that while syndrome specific vulnerable brain networks are targeted in the visual and language variants of AD, all syndromes are commonly affected by DMN (including MTL) degeneration [66]. Our model would therefore greatly benefit from taking into account the patterns of disease spread from the DMN to highly interconnected brain regions, and from incorporating multifocal disease onsets. Finally, we would like to highlight that the proposed model focuses on amyloid-β and tau, the two fundamental pathologies biologically defining the disease. We have hence ignored several co-pathologies associated with AD, especially in older patients and mixed forms of the disease. Additional work is needed to assess how other pathologies—such as TDP-43, Lewy bodies, vascular pathology, or immunological responses—could be integrated in our model.
CONFLICTS OF INTEREST
Alexander Drzezga has equity in Siemens Healthineers, receives lecture fees from Siemens Healthineers, GE Healthcare, AVID/Lilly, Piramal, Life Molecular Imaging, Hennig, and Sanofi, has a patent on PSMA-PET tracer and has current funding from Siemens Healthineers, GE Healthcare, Life Molecular Imaging, and Avid/Lilly. All other authors report no conflicts.
Footnotes
ACKNOWLEDGMENTS
This study is supported by Shahid Beheshti University.