
Abstract
ATP-binding cassette A7 (ABCA7) is a genetic risk factor for late-onset Alzheimer’s disease (AD). It belongs to a group of transporter genes that specializes in regulating lipid transport in the periphery as well as in the brain. ABCA7 has been implicated in a number of roles relating to AD pathology, including phagocytic clearance of amyloid-β peptides. We have discovered that deletion of ABCA7 in mouse causes a dramatic reduction in white adipose tissue (WAT) in female mice. WAT is important in AD context because it is the primary producer of leptin, which is a hormone that is known to modulate AD neuropathology. WAT in male Abca7–/– mice was not altered. The pathological link between ABCA7 and WAT that impacts on AD is unknown. Our transcription analysis revealed that lipin-1 expression was significantly upregulated in female Abca7–/– mice, indicating that ABCA7 affects WAT development. The circulating leptin level was significantly reduced in female Abca7–/– mice without any change in WAT leptin mRNA or protein expression, indicating that ABCA7 does not affect leptin production, but alters the circulating leptin level indirectly by affecting WAT development. Insulin is a key hormone that regulates WAT development, i.e., adipogenesis, and it was significantly reduced in female Abca7–/– mice. These data when put together suggest that ABCA7 plays a role in regulating WAT development and consequently circulating leptin levels, which are known to modulate AD neuropathology.
INTRODUCTION
ABCA7 is a member of the ATP-binding cassette subfamily A (ABCA) that specializes in regulating lipid transport and maintaining lipid homeostasis [1–3]. Recent large genome-wide association and sequencing studies have revealed that ABCA7 is a strong risk factor for late-onset Alzheimer’s disease (AD) [4–7]; ABCA7 is one of top 10 AD susceptibility genes [4, 8]. ABCA7 was first described to play a role in regulating cellular cholesterol and phospholipid transport [2, 3]. Since then, a number of roles relating to AD pathology have been described, including a role in phagocytic clearance of amyloid-β (Aβ) peptides [9–12] and in regulating amyloid-β protein precursor (AβPP) processing to generate Aβ peptides [13–15]. Thus far, evidence suggests that ABCA7 affects multiple pathways impacting AD pathology.
The first study of Abca7 knockout mice revealed that deletion of ABCA7 caused a dramatic reduction in the size of white adipose tissue (WAT) [16]. WAT is important in the AD context because it is the primary producer of leptin, which is known to modulate the levels of AD neuropathology [17–19]. The level of leptin in circulation is dependent on WAT size, i.e., the larger the WAT the higher the leptin level [20, 21]. Leptin is a metabolic hormone that was originally discovered to suppress appetite and regulate energy expenditure [22]. Since its discovery in 1994, leptin has been shown to have other important physiological roles. A substantial body of evidence indicates that leptin regulates a number of features defining AD at the molecular and physiological levels, and that a deficiency in leptin levels may contribute to central nervous system abnormalities leading to AD. Studies have shown that AD patients have significantly decreased circulating leptin levels [23]. A large prospective study of 2,871 subjects demonstrated that elderly individuals, followed over 4 years, with high leptin levels were less likely to develop AD than those with low leptin levels [24]. Another study followed 785 healthy individuals from the Framingham cohort for 8.3 years and showed that higher leptin levels were associated with a lower risk of AD [25]. Therefore, the circulating leptin level is inversely correlated with AD risk, i.e., the higher the leptin level the lower the AD risk.
The pathological link between ABCA7 and WAT that impacts on AD is unknown. We therefore proposed a hypothesis that ABCA7 regulates WAT development and leptin levels and consequently modifies AD neuropathology. In this study we investigate the link between ABCA7 and WAT development, and determine how ABCA7 alters leptinlevels.
MATERIALS AND METHODS
Animals
ABCA7 knockout (Abca7–/–) mice were from Prof Mason Freeman (Massachusetts General Hospital, Boston). They were established on the C57BL/6J background and backcrossed >20 generations. Abca7–/– mice are homozygous for the transgene and the genotype was confirmed by Southern hybridization and polymerase chain reaction (PCR) [16]. Age-matched (∼5 months) wild type C57BL/6J mice were used as non-transgenic controls. Mice were kept in polysulfone cages (2–4 mice/cage) under a 12 : 12 h light:dark schedule with food and water available ad libitum. Each cage provided the same environment and featured a polycarbonate igloo and nesting material. Animal experimental procedures were in accordance with the Australian Code for the Care and Use of Animals in Research and were approved by the Animal Care and Ethics Committee of the University of New South Wales.
Tissue processing
Mice were injected (i.p.) with a lethal dose of pentobarbitone sodium. Blood (∼0.5 ml) was collected from the right heart ventricle and plasma prepared. Transcardial perfusion was performed with phosphate buffered saline (PBS; pH 7.4) through the left heart ventricle. Perigonadal white adipose tissue (WAT) deposits were collected. The left deposit was snap-frozen for subsequent RNA and protein analyses. The right deposit was drop-fixed in the 4% paraformaldehyde (in 0.1 M PBS, pH 7.4) at 4°C for 4 days for histological analysis.
Histology
The fixed WAT was embedded in paraffin as described previously [26]. The 5μm sections were prepared and stained with hematoxylin and eosin (H&E). The five representative photos of each section were taken at 20x magnification with an Axioskop microscope (Zeiss Germany) using AxioCAM Mrc camera. The images were stored in uncompressed 24-bit color TIFF format and analyzed using Image J based Adiposoft software as described previously [27].
RNA extraction and quantitative PCR
RNA was isolated using TRIzol reagent (Invitrogen) following the manufacturer’s protocol. All procedures were carried out using RNase-free reagents and consumables. One microgram of RNA was reverse transcribed into cDNA using Moloney-murine leukemia virus (M-MLV) reverse transcriptase and random primers (Promega, Madison, Wisconsin, USA) in 20μl reaction volume. Quantitative PCR (qPCR) assays were carried out using a Mastercycler ep realplex S (Eppendorf, Sydney, Australia) and the fluorescent dye SYBR Green (Bio-Rad), following the manufacturer’s protocol. Briefly, each reaction (20μl) contained 1x mastermix, 5 pmoles of primers and 1μl of cDNA template. Amplification was carried out with 40 cycles of 94°C for 15 s and 60°C for 1 min. Gene expression was normalized to the housekeeper gene GAPDH. A no-template control was included for each PCR amplification assay. The level of expression for each gene was calculated using the comparative threshold cycle (Ct) value method using the formula 2–ΔΔCt (where ΔΔCt =ΔCt sample –ΔCtreference).
ELISA
Plasma leptin and insulin were measured by ELISA following manufacturer’s protocols –leptin (Millipore cat.# EZML-82K) and insulin (Millipore cat.# EZRMI-13K). WAT leptin was also measured by ELISA. Protein lysates were prepared from WAT by homogenization of 100 mg of tissue in chilled sucrose buffer (250 mM sucrose, 10 mM HEPES pH 7.4, 1 mM EDTA) supplemented with protease inhibitor cocktail (Roche), followed by centrifugation at 1000 g for 10 minat 4°C.
Cell culturing and adipocyte differentiation
3T3-L1 preadipocytes were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 100 IU/ml penicillin and 100μg/ml streptomycin at 37°C in humidified air containing 5% CO2. The 3T3-L1 cells were differentiated into adipocytes as described previously [28]. Briefly, they were grown to 90% confluence and treated with 0.5 mM methyl-isobutylxanthine (Sigma I17018), 1μM dexamethasone (Sigma D4902) and 5μg/ml insulin (Sigma I0516). After 2 days the media was replaced with DMEM supplemented with 10% FBS and 5μg/ml insulin. Thereafter, the media was replaced every two days for ∼10 days. Differentiated adipocytes were confirmed by Oil O Red staining of the lipid pools present in the adipocytes. Briefly, the cells were washed with PBS, fixed with 10% formalin in PBS for 2 min and washed with distilled water. The cells were then stained with Oil O Red (Sigma O0625) for 1 hour, washed twice with distilled water and examined under a lightmicroscope.
Transfection
Transient transfection was performed using Lipofectamine 2000 and Opti-MEM-I (Invitrogen) following the manufacturer’s protocol. Firstly, the 3T3-L1 cells were cultured in 12-well plates and differentiated into adipocytes as described above. ABCA7 cDNA-lipofectamine complex was then added to the cells in an antibiotic-free media, and after 24 h the cells were harvested for gene expression analysis.
Statistical analysis
Data presented are expressed as mean±SE shown by the error bars. Statistical significance was determined using Student’s t-test with a p value < 0.05 considered significant.
RESULTS
Deletion of ABCA7 causes a reduction in white adipose tissue in female Abca7–/– mice
We have previously reported that deletion of ABCA7 causes a significant reduction in WAT mass in female mice [16]. The difference is quite striking with ∼40% less WAT in female Abca7–/– mice compared to littermate wild type female mice (Fig. 1A). The size of WAT was not altered in male Abca7–/– mice. We have also measured the weight of liver and found no significant difference (data not shown). To determine whether the reduction was due to changes in adipocytes, the predominant cells that make up WAT, we prepared and analyzed histological sections of WAT from Abca7–/– and wild type mice. We found no significant difference in the morphology (Fig. 1B) or cell size between Abca7–/– and wild type mice of both sex (Fig. 1C).
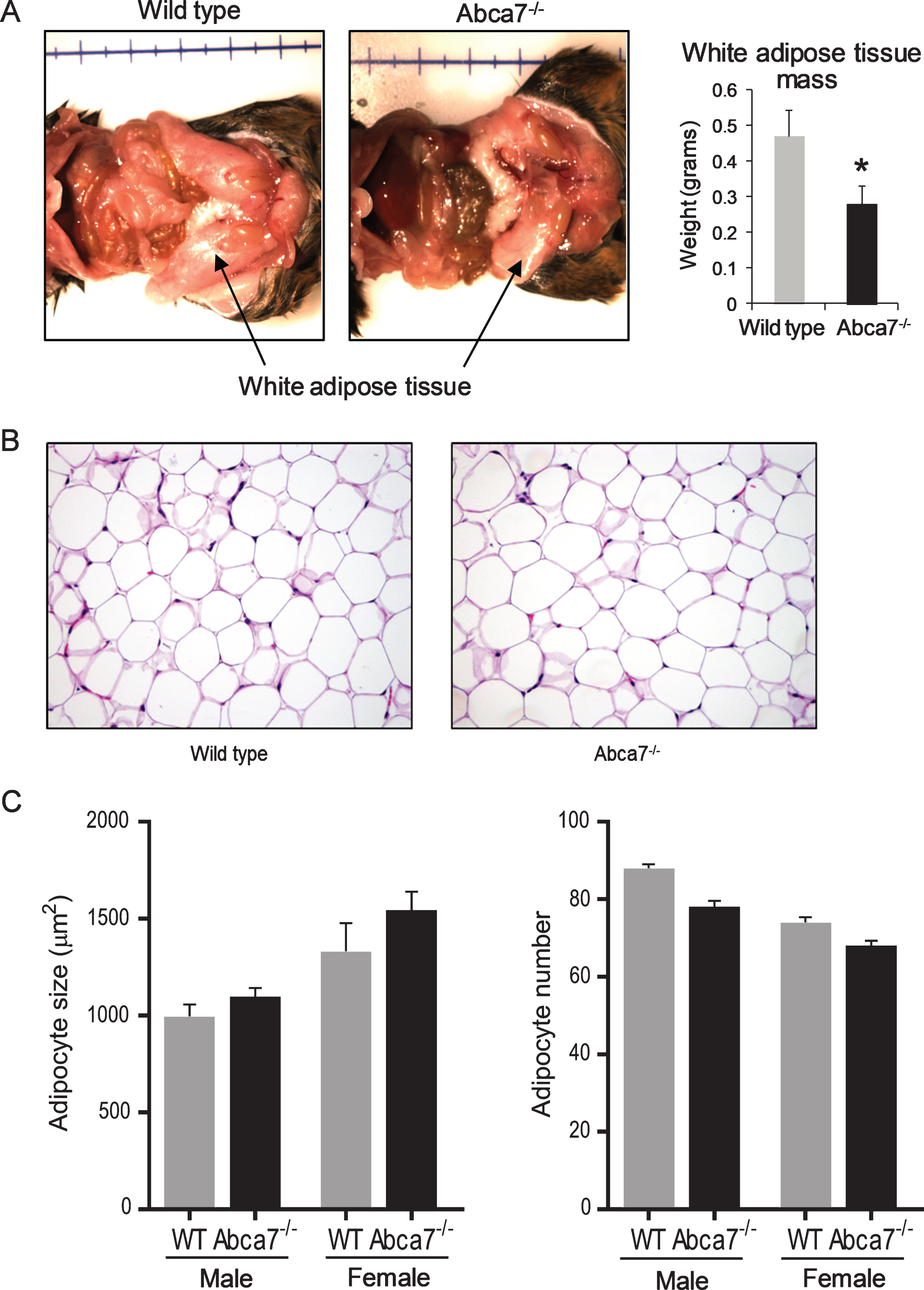
Deletion of ABCA7 causes a reduction in white adipose tissue (WAT) in female Abca7–/– mice. A) WAT mass is dramatically reduced in female Abca7–/– mice. Data represent mean (n = 13) and SE as error bars, *p < 0.05. B) A representative H&E stained section of WAT from female wild type and Abca7–/– mice. C) No significant difference in adipocyte cell size or number between wild type and Abca7–/– mice. Data represent mean (n = 6) and SE as error bars.
Lipin-1 transcription is upregulated in white adipose tissue of female Abca7–/– mice
To understand the changes in WAT caused by ABCA7 deletion, we compared global mRNA expression of WAT from Abca7–/– and wild type mice. Our specific question was does ABCA7 affect WAT development. We discovered that lipin-1 was upregulated by 1.87 fold (p < 0.0001) in female Abca7–/– mice. This data was subsequently confirmed by qPCR (Fig. 2A). The lipin-1 expression was unaltered in male Abca7–/– mice (Fig. 2A). WAT triglyceride level was also significantly decreased in female Abca7–/– mice (Fig. 2B). Lipin-1 encodes the enzyme phosphatidate phosphatase that catalyses the conversion of phosphatidate to diglyceride, which is a key step in triglyceride production in WAT (Fig. 2C). Triglyceride is a major lipid in the lipid pools of adipocytes in WAT [29]. WAT development is dependent on triglyceride production in WAT. Lipin-1 deficiency causes generalized lipodystrophy and impaired WAT development [30]. Upregulation of lipin-1 transcription would indicate lipin-1 enzyme deficiency or dysfunction in female Abca7–/– mice. We therefore provide evidence that ABCA7 affects WAT development.
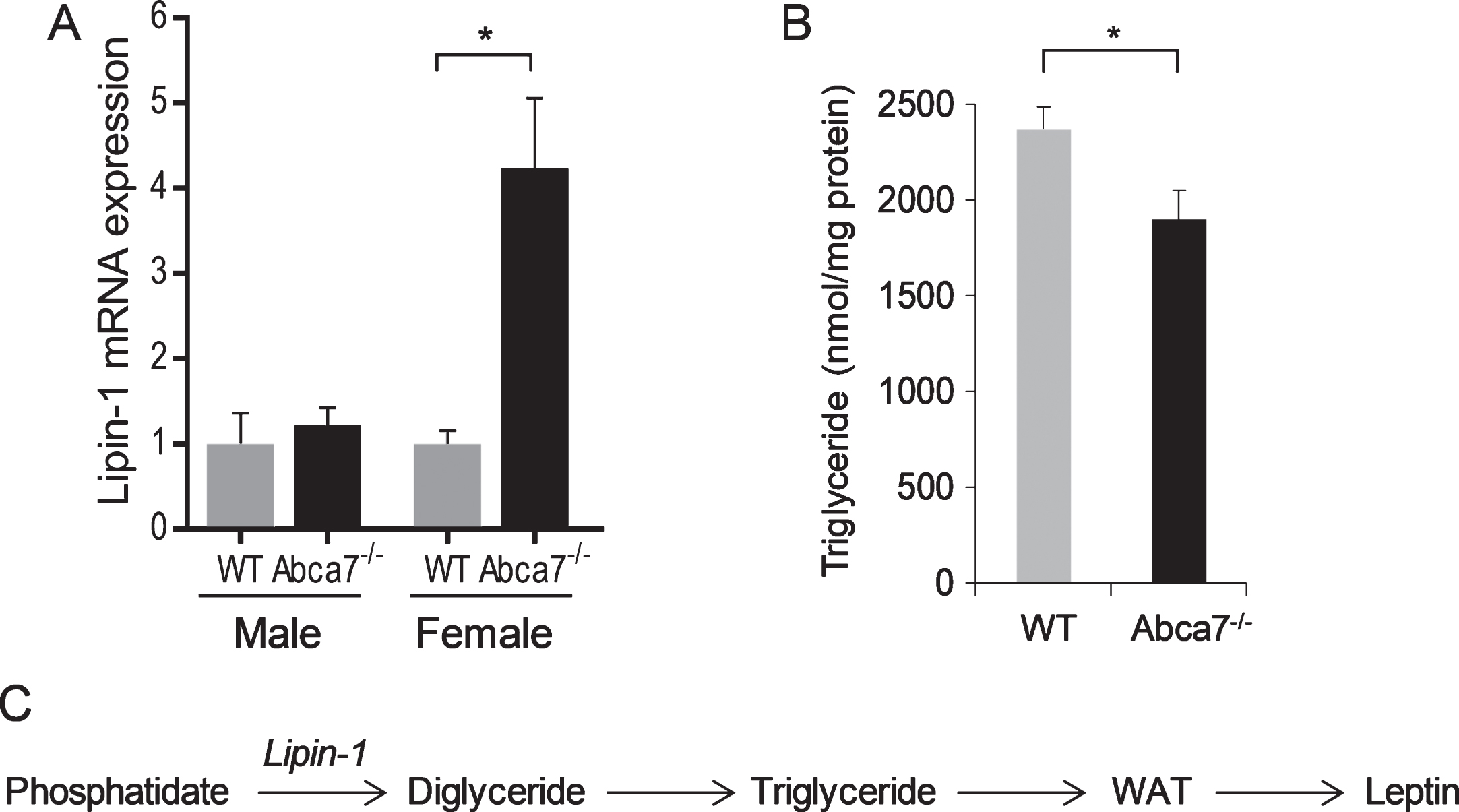
Lipin-1 expression in white adipose tissue. A) Lipin-1 mRNA expression in WAT of wild type and Abca7–/– mice as measured by qPCR. Data represent mean (n = 10) and SE as error bars, *p < 0.05. B) WAT triglyceride level is significantly decreased in female Abca7–/– mice. Data represent mean (n = 6) and SE as error bars, *p < 0.05. C) Lipin-1 converts phosphatidate to diglyceride and triglyceride, which is essential for WAT development and leptin production.
Deletion of ABCA7 reduces the circulating leptin level
Since leptin is primarily produced by WAT and the fact that the circulating leptin level is directly proportional to WAT size [20, 21], i.e., the larger the WAT the higher the leptin level (Fig. 3A), we were interested if the circulating leptin level is altered with ABCA7 deletion. As mentioned above, the circulating leptin level is important in the AD context [23, 31], i.e., the higher the leptin level the lower the AD risk (Fig. 3B). Here, we measured the circulating leptin level of Abca7–/– and wild type mice of both sex. Blood was collected (by cardiocentesis) from animals that were fasted overnight and plasma prepared and leptin measured by ELISA. We found that the circulating leptin level was significantly reduced in female Abca7–/– mice compared to female wild type mice (Fig. 3C). The circulating leptin level was not significantly altered in male Abca7–/– mice (Fig. 3C).
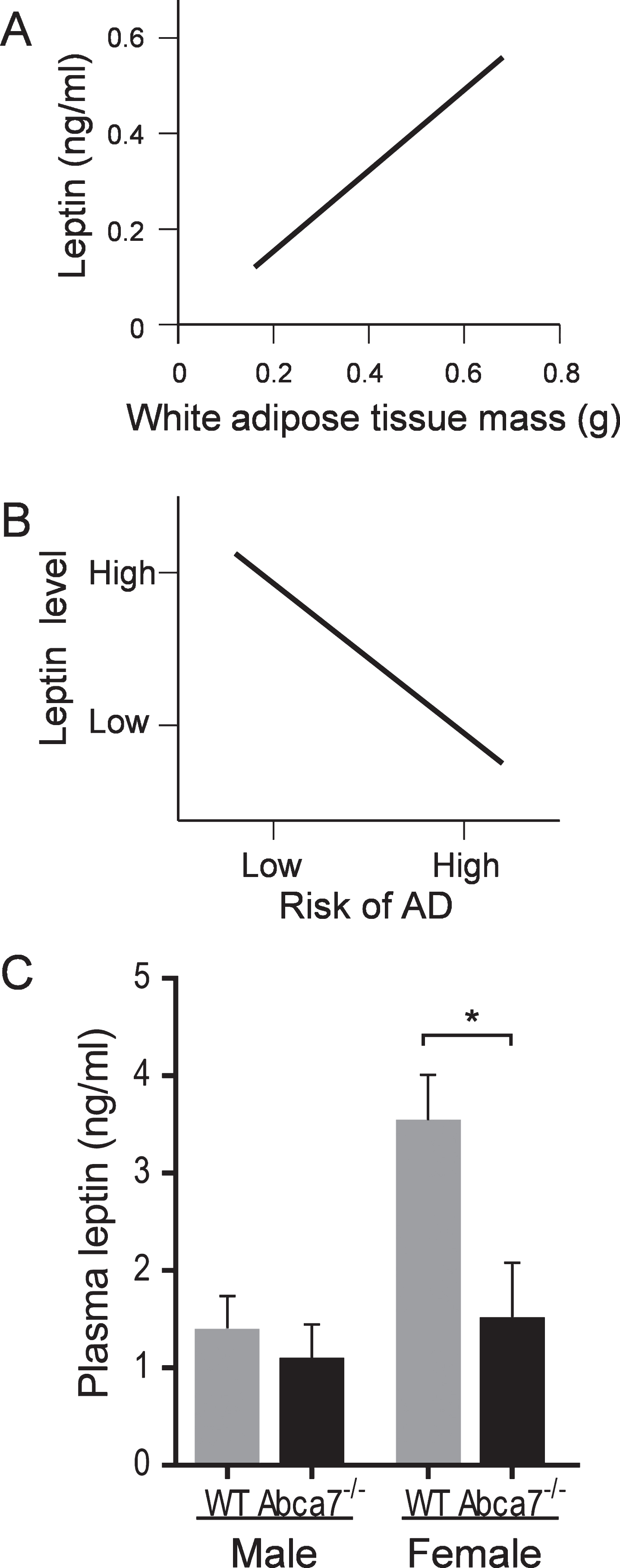
Deletion of ABCA7 reduces the circulating leptin level in female Abca7–/– mice. A) A theoretical plot depicts the linear relationship between circulating leptin level and WAT mass. B) A theoretical plot depicts the inverse relationship between circulating leptin level and AD risk, i.e., the higher the leptin level the lower the AD risk. C) Plasma leptin levels were significantly reduced in female Abca7–/– mice as measured by ELISA. Data represent mean (n = 6) and SE as error bars, *p < 0.05.
Leptin expression in white adipose tissue is unaltered with ABCA7 deletion
We also measured leptin mRNA and protein expression in WAT to determine whether ABCA7 directly affects leptin production. We prepared mRNA and protein from the same quantity of WAT of Abca7–/– and wild type mice of both sex and analyzed leptin expression by qPCR and ELISA. We found no significant difference in either mRNA or protein expression between Abca7–/– and wild type mice of both sex (Fig. 4A). We also conducted an in vitro study to test if ABCA7 had any impact on leptin expression. We differentiated 3T3-L1 preadipocytes into adipocytes; the expression of ABCA7 is upregulated in adipocytes (Fig. 4B). We then transfected adipocytes with ABCA7 cDNA and measured leptin expression. Despite the significant increases in ABCA7 expression (Fig. 4C), we found no significant change in leptin mRNA expression (Fig. 4D). These data indicate that ABCA7 regulates the circulating leptin level indirectly by altering WAT size rather than altering WAT leptin expression.
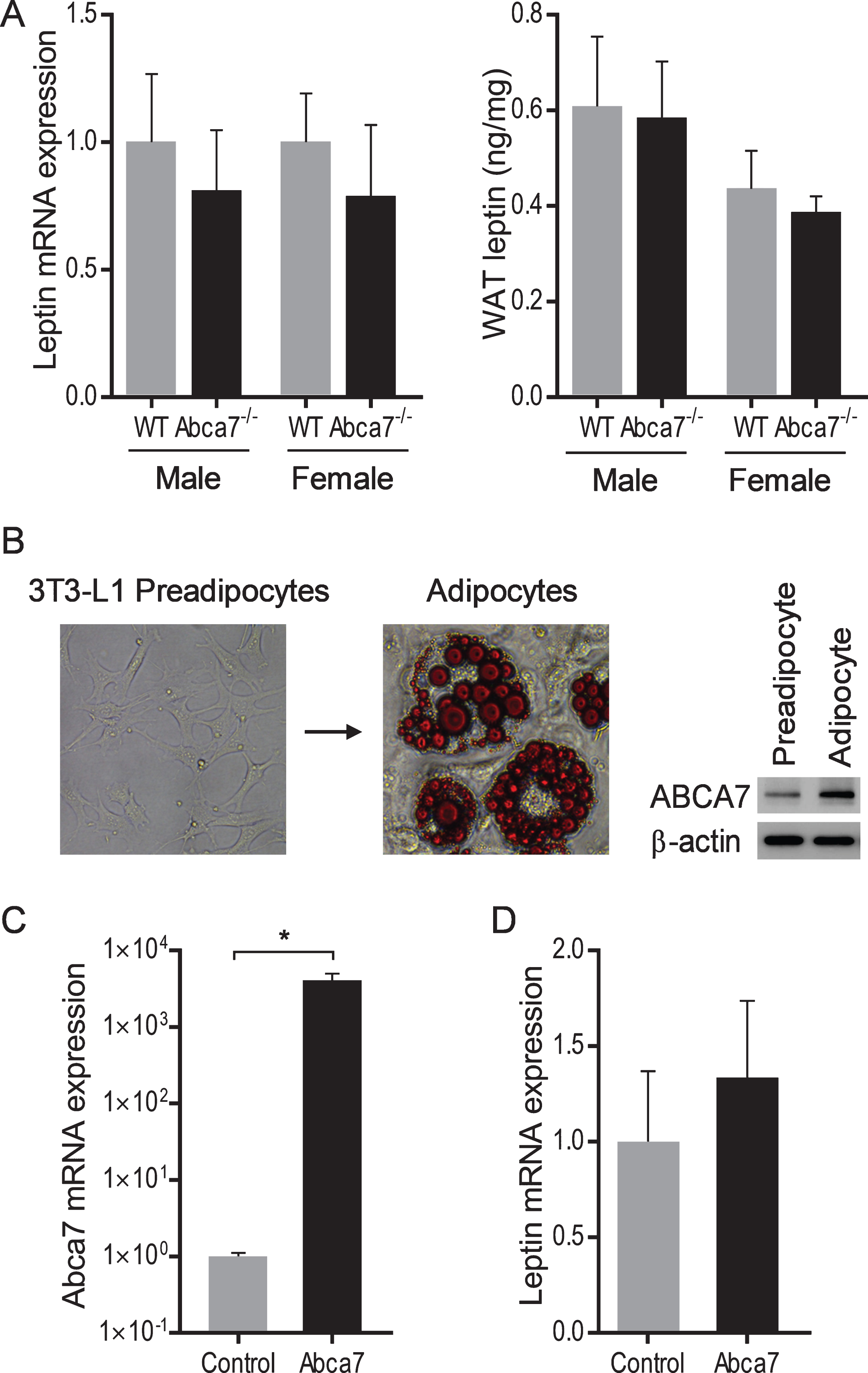
Leptin expression in white adipose tissue and adipocytes. A) No significant difference in leptin mRNA or protein expression in WAT between wild type and Abca7–/– mice as measured by qPCR and ELISA respectively. Data represent mean (n = 6) and SE as error bars. B) 3T3-L1 preadipocytes were differentiated into adipocytes. Fully differentiated adipocytes contain lipid pools as stained by Oil O Red. The expression of ABCA7 is upregulated in adipocytes. C) Adipocytes were successfully transfected with Abca7 cDNA as measured by qPCR; an empty vector was used as a control. Data represent mean (n = 6) and SE as error bars, *p < 0.01. D) No significant change in leptin mRNA expression in the Abca7-transfected adipocytes as measured by qPCR.
Deletion of ABCA7 reduces the circulating insulin level
Since insulin is recognized as a key hormone that regulates WAT development, i.e., adipogenesis [32–36], we were interested if the circulating insulin level is altered with ABCA7 deletion. Using the same plasma samples prepared from Abca7–/– and wild type mice above, we measured the circulating insulin levels by ELISA. We found that the circulating insulin levels were significantly reduced in female Abca7–/– mice compared to female wild type mice (Fig. 5). The circulating insulin levels were not significantly altered in male Abca7–/– mice (Fig. 5). These data suggest a plausible explanation that ABCA7 deletion impacts on circulating insulin levels leading to decreased WAT development in female mice.
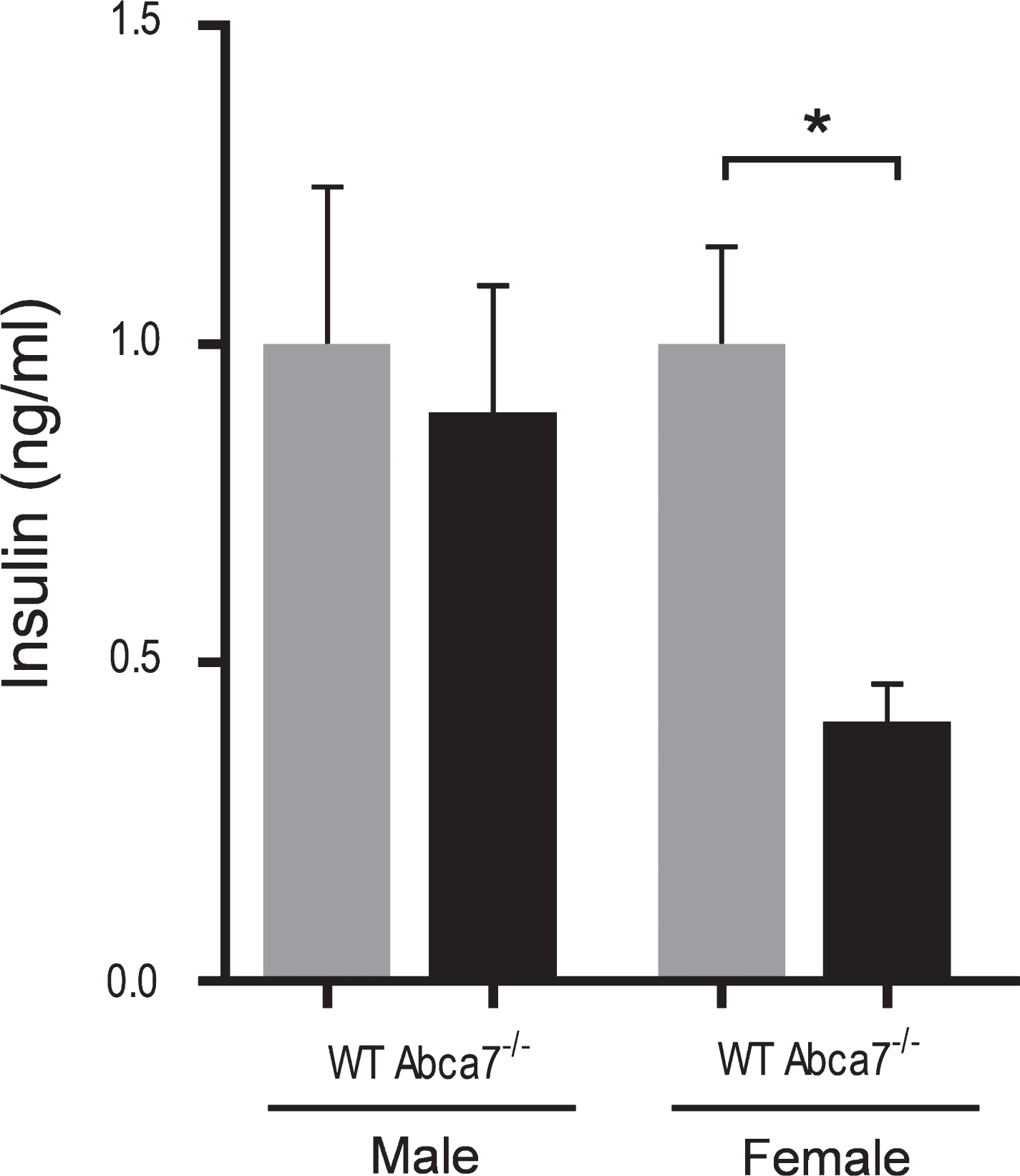
Deletion of ABCA7 reduces the circulating insulin level. Plasma insulin levels were significantly reduced in female Abca7–/– mice as measured by ELISA. Data represent mean (n = 6) and SE as error bars, *p < 0.05.
DISCUSSION
ABCA7 is a member of ABCA subfamily that predominantly regulates the transport of lipids and other lipophilic molecules across membranes [37, 38]. Genome wide association studies have identified ABCA7 as a strong susceptibility gene for AD [4–6]. Previous studies have shown that deletion of ABCA7 in a mouse model of AD exacerbates cerebral Aβ neuropathology [12], and causes reduced microglial phagocytic clearance of Aβ peptides [11]. Apart from the effect on Aβ neuropathology, ABCA7 deletion causes a reduction in WAT in female mice [16]. The size of WAT in the AD context is important because the circulating level of leptin produced by WAT is directly proportional to WAT size, and higher circulating leptin levels reduce the risk of developing AD [23–25]. We have shown that the circulating leptin level is significantly decreased in female Abca7–/– mice, consistent with their reduced WAT size. The size of WAT and circulating leptin levels were unaltered in male Abca7–/– mice. We have also shown, using in vivo and in vitro analyses, that ABCA7 does not alter leptin mRNA or protein expression, indicating that the level of leptin produced per unit of WAT mass is unaltered in Abca7–/– mice. Therefore, ABCA7 regulates the circulating leptin level indirectly by altering WAT size rather than altering WAT leptin production.
Human clinical and epidemiological studies have overwhelmingly demonstrated that leptin is strongly associated with cognition and memory formation and that circulating leptin levels are inversely correlated with AD risk [23–25, 31]. Furthermore, administration of leptin results in significant improvements in cognition in leptin-deficient patients [39] and in transgenic animal models of AD [17]. In vitro studies show that leptin reduces Aβ secretion [19]. In animal models of AD, leptin reduces soluble Aβ levels and Aβ plaque load in the hippocampus [17]. Further evidence of the involvement of leptin in AD pathogenesis comes from the fact that leptin receptors are highly expressed in the hippocampus [40, 41], a region of the brain intrinsically involved in memory and cognition. In hippocampal CA1 neurons, leptin facilitates presynaptic transmitter release and postsynaptic sensitivity, resulting in improved spatial learning and memory [42]. These data strongly suggest that leptin has the potential to modulate AD pathogenesis and reinforces the emerging consensus that leptin homeostasis could be a possible therapeutic target for AD treatment.
We have shown that lipin-1 transcription in WAT is significantly upregulated in female Abca7–/– mice, providing evidence that ABCA7 affects WAT development. Lipin-1 is essential for the production of triglyceride required for WAT development [30]; triglyceride is a major lipid in the lipid pools of adipocytes in WAT [29]. Uncontrolled overexpression of lipin-1 protein would lead to obesity [43]. Upregulation of lipin-1 transcription would indicate that that lipin-1 is deficient or dysfunctional, resulting in impaired WAT development, which is the case for female Abca7–/– mice. It is interesting to note that lipin-1 has a secondary function in that it regulates extracellular signal-regulated kinase (ERK) in skeletal muscle differentiation [44]. Previously, it has been shown that ERK phosphorylation process was defective in ABCA7-deficient cells [45]. It is also possible that other factors may have influenced the expression of lipin-1, such as stress; stress would cause increases in glucocorticoid levels, which in turn upregulates lipin-1 expression [46]. A detailed analysis of lipid content of WAT in Abca7–/– mice would help to further understand the role of ABCA7 in adipogenesis and WAT development.
We showed that the circulating insulin level was severely affected by ABCA7 deletion. Insulin is a master regulator of adipogenesis, the process of differentiating preadipocytes into mature adipocytes [32]. Therefore, ABCA7 appears to affect WAT development, i.e., adipogenesis, via altering the circulating insulin level. The development of fully differentiated adipocytes from preadipocytes requires a cascade of transcriptional factors, which are activated by insulin. The transcription factors are critical for attaining the mature morphological and biochemical phenotype of adipocytes [33, 34]. Insulin is also known to activate lipogenic and glycolytic enzymes that are required for lipogenesis in WAT development [36], and therefore other roles of insulin in WAT development could also be affected by ABCA7 deletion.
At this stage it is unclear why there is a gender difference in the effect of ABCA7 deletion on insulin levels and WAT development. WAT development is a complex process that requires a number of hormones including sex steroids [47]. Whether ABCA7 affects the level of sex steroids is yet to be determined. What is interesting is that sex steroids (i.e., estrogens and testosterone) are synthesized from the precursor molecule cholesterol. Since ABCA7 is a regulator of cholesterol metabolism, it is plausible to think that ABCA7 deletion could affect sex steroid production. Changes in cholesterol level in the gonads impact on sex steroid production [48]. In the case of ABCA1, a member of the ABCA subfamily and a homolog of ABCA7 [3], the location of insulin regulation appears to be at the pancreas [49–51]. ABCA1 deletion or loss-of-function mutations cause accumulation of cholesterol in cells resulting in decreases in high density lipoprotein (HDL)-cholesterol [52]. ABCA1 deletion in mice causes accumulation of cholesterol in the β cells of the pancreas causing impaired insulin secretion [49, 50]. Likewise in humans, loss-of-function mutations in ABCA1 cause reduction in the insulin secretory capacity of the β cells of the pancreas [51]. It is interesting to note that ABCA7 deletion also causes decreases in HDL-cholesterol but only in female Abca7–/– mice [16]. Whether the insulin secretory capacity of the pancreas is impaired in female Abca7–/– mice is yet to be determined.
There is a considerable amount of data which indicate that ABCA7 contributes to AD pathology [12, 14, 53–59], particularly affecting AβPP processing [14, 53] and the phagocytic clearance of Aβ peptides [45, 60–62]. Our current study is the first report indicating that ABCA7 alters WAT development. This study has revealed a pathway by which ABCA7 regulates WAT development and leptin levels. It has identified new targets for controlling leptin levels that may impact AD pathology. Leptin has not been used in clinical trial to treat AD and therefore this work provides an invaluable preclinical proof-of-concept that leptin is a potential therapy for AD and for those at risk presenting with low leptin levels.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Footnotes
ACKNOWLEDGMENTS
This work was supported by grants from the Mason Foundation (to W.S.K.) and the University of New South Wales Goldstar Award (to W.S.K.). G.M.H. is a National Health and Medical Research Council (NHMRC) Senior Principal Research Fellow and S.B. is a NHMRC Research Fellow. The authors wish to thank Prof M. Freeman (Massachusetts General Hospital, Boston) for Abca7–/– mice.