
Abstract
The aim of this review is to examine the causal mechanisms underlying the relationship between MDD and CHD, with the aim of constructing a topological map of the causal network which describes the relationship between MDD and CHD.
Keywords
Depression and coronary heart disease (CHD) are important global public health problems. Depression is one of the largest causes of morbidity worldwide and contributing heavily to estimates of global burden of disease [1]. Major depressive disorder (MDD) is also a significant problem in people with co-morbid medical illness. An average of between 9.3% and 23% of individuals with one or more chronic physical disease has co-morbid depression [2]. Similarly, CHD causes a significant burden of disease and is a leading cause of mortality and morbidity worldwide. The worldwide prevalence of angina pectoris (a readily identifiable illness related to CHD) is 54 million people worldwide [3].
It is well established that there is a link between MDD and CHD [4–9]. Additionally, depression and CHD have a complex reciprocal interrelationship, where depressive illness leads to ischaemic heart disease, and heart disease leads to depression [10,11]. Depression has been linked with a 3- to 4-fold increase in the risk of recurrent cardiac events as well as death [12]. Depression also increases the risk of future cardiac mortality and morbidity in patients with coronary artery disease [13,14]. Conversely, people with CHD have a prevalence of depression approximately three times higher than healthy people [15]. Thombs et al. [16] found that major depression is also three times more common in patients after an acute myocardial infarction (AMI) than in the general population.
Objective
If links between CHD and MDD exist, it raises the possibility that common mechanisms could be identified which link these diseases. Moreover, this knowledge could then be applied to improve prevention and treatment for both conditions. However, despite strong evidence for a link between depression and CHD, there is no single comprehensive model that describes the mechanism for this relationship [17]. Several individual pathways have been described. These pathways have typically been discussed as separate entities in empirical research and reviews have described a small number of related mechanisms in detail. For example, both Tracey [18] and Musselman et al. [7] provide excellent reviews of the relationship between autonomic mechanisms and inflammatory mechanisms linking MDD and CHD. However, there is considerable overlap and inter-connectivity between all known mechanisms, to the point where these previously discretely described causal mechanisms should be seen as a network of pathogenic determinants.
De Jonge et al. (in press) reviewed the possible mechanisms by which MDD and CHD may be related [19]. The authors identified the relationship between MDD and CHD as a complex system, or network, and suggested that the network might be studied using systems biology. This idea was not further explored however. While the paper offers a lucid review of the literature around the causal pathways linking MDD and CHD, it did not provide a topological map of this network or suggest how systems biology might be applied to it. It also does not adequately address the behavioural mechanisms linking MDD and CHD, which forms and integral component of the overall relationship of MDD and CHD.
The aim of this review is to present the key mechanisms linking major depression and CHD that have been described in the literature, with the aim of constructing a topological map of the causal network which describes the relationship between MDD and CHD. This review will also explore the concept that systems biology, which deals with the study of biological networks, can provide a useful framework to investigate the relationship between the two disease entities.
The relationship between MDD and CHD has also been discussed in the context of other mental disorders, such as anxiety disorders, or personality and character traits (e.g. Rozanski et al. [15]). While the study of such relationships may yield further proposed mechanisms which contribute to the causal network between MDD and CHD, they fall beyond the scope of this review.
Method
Study selection
The search term ‘depression and heart disease’ was entered into an electronic multiple database search engine, ‘Griffith University Library Multiple Database Search’, which searched the following databases: MEDLINE 1950 to present (Ovid), BioMed Central, BMJ Clinical Evidence, BMJ Journals Online, Cambridge Journals Online, Highwire Press, IngentaConnect, LANGE Educational Library, Lippincott, Williams & Wilkins e-journals (Ovid), LocatorPlus, MediLexicon, MEDLINE (EBSCOhost), MEDLINE (via PubMed), MEDLINE – OLDMEDLINE 1951-1965 (Ovid), MEDLINE In-Process (Ovid), Oxford Journals Online, PLoS: Public Library of Science, ProQuest (Multiple Databases), ProQuest Dissertations and Theses – Full Text, PsycINFO (Ovid), PubMed, ScienceDirect, Science Direct: Medicine and Dentistry, Scirus (Elsevier), SpringerLink, STAT!Ref: NURSING: Allied Health Collection, SUMSearch, SwetsWise, Taylor and Francis eBookstore, Web of Knowledge (ISI), Web of Science (ISI) and Wiley InterScience. No constraints were applied to the search. Abstracts were screened for relevance and individually selected articles were then collated. The reference lists of relevant articles were then screened for further articles of interest. Articles were selected on the basis of (i) specifically addressing causal mechanisms, (ii) including discussions of aetiology that included common behaviours, genetics and/or physiological abnormalities or medical co morbidity between the diagnoses, and (iii) those articles specifically addressing the outcomes, prognosis and/or treatment related issues linking depression and cardiac disease.
Results
Mechanisms which link CHD and depression
Six principal mechanisms have been put forward to explain the link between major depression and CHD: (i) behavioural mechanisms, (ii) genetic mechanisms, (iii) dysregulation of immune mechanisms, (iv) coagulation abnormalities and vascular endothelial dysfunction, (v) polyunsaturated omega-3 free fatty acid deficiency, and (vi) autonomic mechanisms. Other mechanisms have been implicated in the relationship between MDD and CHD, but have little supporting evidence to date. For example, antidepressant cardiotoxicity is suggested to account for the increased incidence of adverse cardiac events in depressed people [20]. However, this is unlikely to represent a significant mechanism. The observed relationship between MDD and CHD precedes the development of antidepressant medications, selective serotonin re-uptake inhibitors (SSRIs), which are now the first line of pharmacological therapy for depression, have few cardiotoxic side effects, and the cardiotoxic reactions to antidepressants are generally not severe or life threatening [20].
Although the newer generation of antidepressants has many advantages from a cardiac perspective over tricyclics, the role of antidepressant cardiotoxicity as a confounding variable cannot be excluded here as SSRIs may also have significant cardiac effects. These have been reviewed by Pacher and Kecskemeti who point out the significant effects of SSRIs clinically in reports where they have been shown to produce bradycardia, QT prolongation, decreased T wave amplitude, dysrthymia, syncope and falls, and effects on the vasomotor centre and blood pressure [21]. They have also been shown to affect cellular physiology in vitro with effects on cardiac action potentials and on cardiac ion channels.
Behavioural mechanisms
Depressed people tend to engage in poor health behaviours such as smoking, low physical activity and maintaining a poor diet [15,17]. As a result they have increased rates of obesity and diabetes, which are risk factors for cardiac disease [22]. Depression has been identified as a risk factor for diabetes [23–25] and diabetes in turn is a risk factor for CHD [26]. Depression is also linked to increased rates of obesity [27,28]. In a large prospective cohort study, where 2088 people were followed up over 5 years, depressed participants showed a significantly greater increase in abdominal obesity, especially in visceral fat, than non-depressed people [29]. A meta-analysis of 15 studies on the relationship between obesity and MDD confirmed a reciprocal link between depression and obesity [30]. Obesity was thus found to increase the risk of depression, but additionally, depression was found to be a predictor for developing obesity. Links between obesity and sympathetic autonomic dysfunction in depression are discussed in Autonomic mechanisms below.
Metabolic syndrome is a combination of metabolic conditions which increase the risk of developing cardiovascular disease and insulin resistance. Conditions associated with metabolic syndrome, namely obesity, diabetes, and hyperglycaemia, have also been associated with the presence of depression [31]. In both men and women, metabolic syndrome has been associated with an increased prevalence of depression [32]. This association between metabolic syndrome and depressive illness was independent of age, smoking status, socio-economic factors, and lifestyle. Metabolic syndrome, in turn, has been associated with significantly increased risk of all cause and cardiovascular morbidity and mortality [33–35].
In addition, depressed people have maladaptive coping styles, suffer from chronic life stresses and have increased rates of social isolation and poorer social skills [36–38]. Depressed patients are also less likely to adhere to medical treatment, contributing to poor health outcomes [20,39,40].
Social isolation in particular has been shown to contribute significantly to poor cardiac health and even increased cardiac mortality [41]. While objectively poor social support networks are associated with a two- to three-fold increase in the incidence of CHD over time, low levels of perceived emotional support by the patient also result in an increased risk of future adverse cardiac events [15]. An almost three-fold increase in subsequent adverse cardiac events has been found in patients post-MI who had low levels of emotional support [42]. The direction of causality between social support and depression is still not clear, however, even though these variables have been shown to influence each other [43]. Inadequate social support and social isolation may lead to depression, particularly in the context of a significant stressor such as an MI [44].
Psychological distress, low self-esteem and impairment in occupational and social functioning occurs in patients after MI [45,46]. While psychological distress in such studies is discussed along with depressive illness and anxiety symptoms, the question whether stress and distress following an MI then lead to further deterioration in the form of MDD is not entirely clear. It could be argued, however, that factors such as distress around loss of function post-MI, for example, combined with factors such as limited social support, could directly contribute to an increased risk of MDD.
Depressed patients have higher rates of daily smoking and nicotine dependence [47,48] and are less likely to give up smoking [49–52]. Cigarette smokers have almost three times the rate of acute MI as compared with non-smokers [53], potentially establishing a causal link between increased smoking behaviour in MDD and the increased incidence of CHD.
The behavioural mechanisms are summarized in Figure 1. Despite the above evidence, however, there is an increase in cardiac disease risk in depressed patients even after cardiac risk factors such as smoking are statistically adjusted for. Thus, while behavioural factors play a role in increasing the risk of cardiac disease in depressed patients, they do not on their own account for the link between the two disease entities.
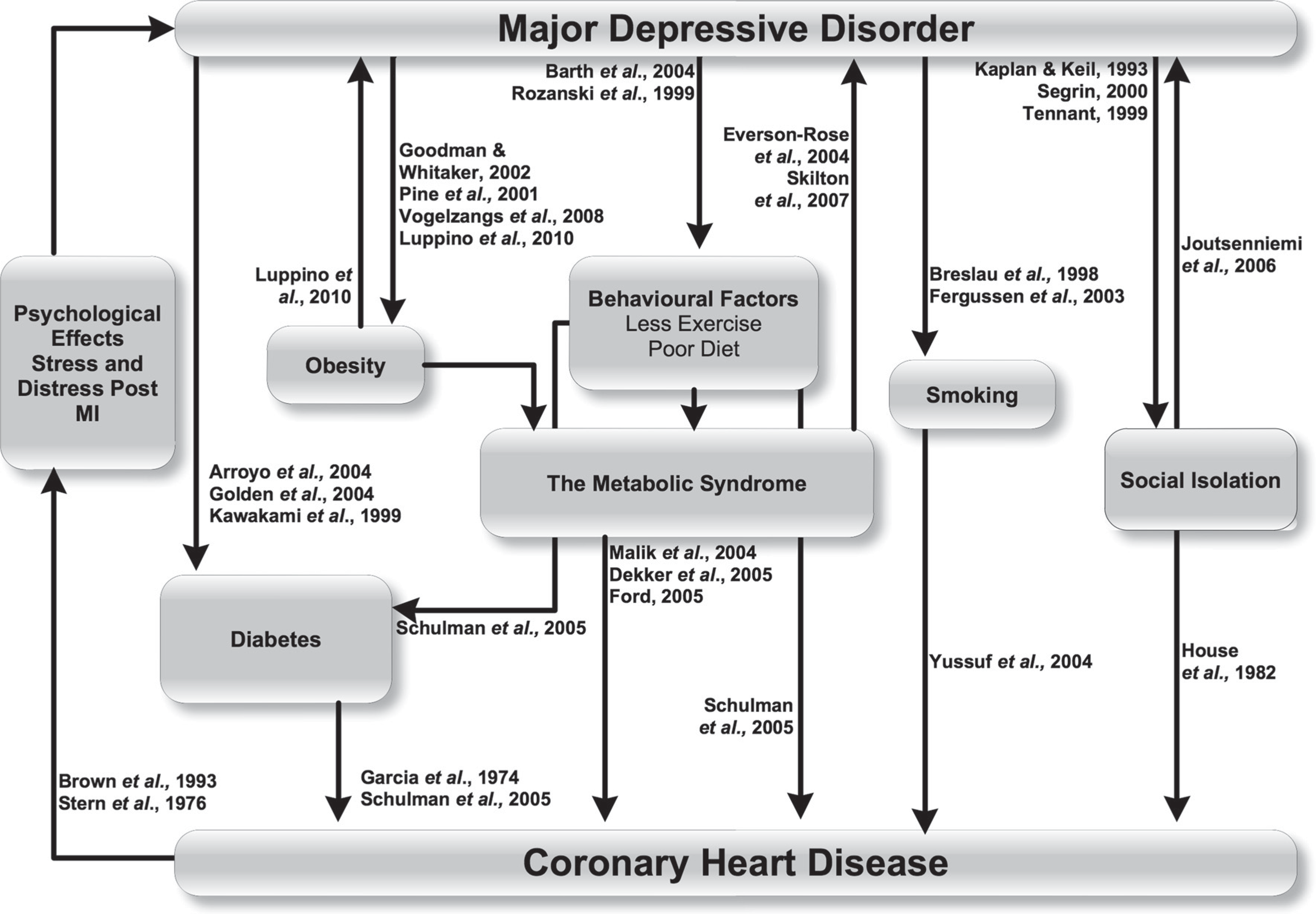
Behavioural mechanisms linking major depressive disorder and coronary heart disease.
Genetic mechanisms
There is some evidence to support genetic mechanisms which link MDD and CHD. Despite some promising findings [54], the genetic mechanisms linking depression and cardiovascular disease are complex and this field still requires much further research. While a comprehensive examination is beyond the scope of this review, some of the pertinent findings are discussed below.
The 5-HTT serotonin transporter gene codes for the serotonin transporter protein, which facilitates the re-uptake of serotonin from the synaptic cleft in serotonergic neurons. Genetic polymorphism of the serotonin transporter gene has been implicated in the pathogenesis of MDD [55–57]. People who have one or two short alleles of the 5-HTT serotonin transporter gene become depressed more often after stressful events than people who have two long alleles of this gene [58]. The short allele of this polymorphism can reduce the transcription efficiency of the gene, resulting in reduced expression of the serotonin transporter in the membrane of the pre-synaptic neuron. The short allele has been shown to predict depression in the presence of stressful life events [58–65], although the mechanism by which it results in MDD is still not known [66]. There is also evidence of the short allele being associated with MDD in patients after suffering an acute coronary syndrome [67]. A possible link also exists between the short allele of the serotonin transporter genome and sympathetic nervous activation. Otte et al. [66] demonstrated that among people who were carriers of the short allele and who had stable CHD, there was an increased risk of depression, an increase in perceived stress levels, as well as increased secretion of noradrenaline. Increased sympathetic activation in turn has been linked to CHD, as discussed in the section on Autonomic mechanisms below.
The serotonin transporter protein (5-HTT) is also the site of action of antidepressants which block serotonin re-uptake from the synapse [68,69]. Common genetic polymorphisms of several monoamine systems, such as 5-HTT, MAO-A or COMT, have been studied as candidate genes for mood disorders [70]. However, it has also been proposed that the serotonin transporter gene functional polymorphism may confer susceptibility for some cardiovascular risk factors as well as for mood disorders [70]. The hypothesis has thus been proposed that serotonin transporter gene functional polymorphism could also represent a mechanism for depression-related increased cardiovascular morbidity and mortality [70]. There is, however, little direct evidence of the genetic contribution to depressive symptoms with CVD [54]. A candidate gene study on French-Canadian subjects with established CVD focused on genes related to inflammation, platelet aggregation, endothelial function and omega-3 fatty acid metabolism as predictors of depressive symptoms [54]. The study found that genetic variation in genes relevant to endothelial dysfunction and platelet aggregation could contribute to depression in cardiac patients. This study thus relates possible genetic mechanisms to inflammatory mechanisms, endothelial dysfunction and platelet activation.
The relationships between these mechanisms are shown in Figure 2.
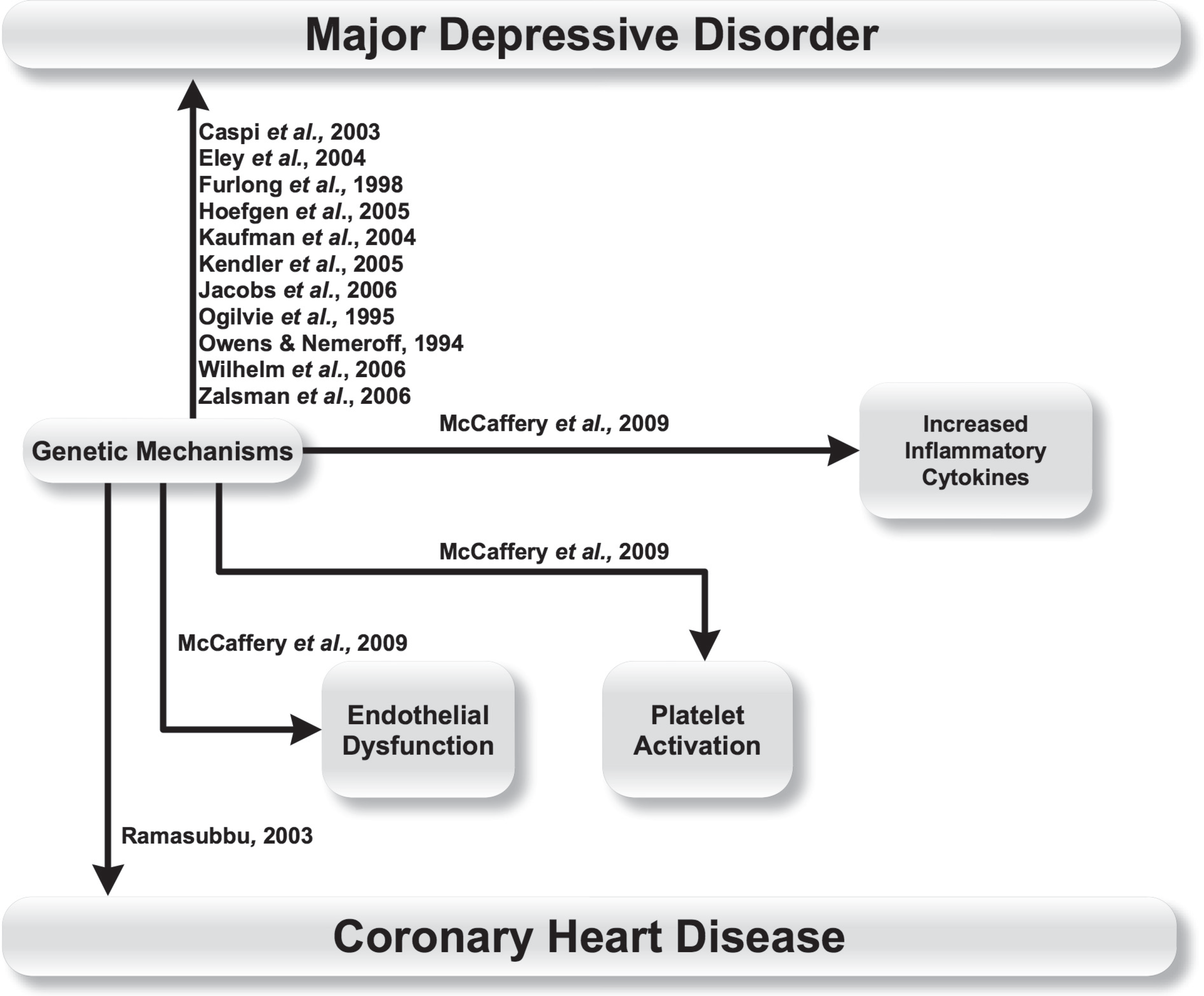
Genetic mechanisms linking major depressive disorder and coronary heart disease.
Inflammatory mechanisms
Dysregulation of immune mechanisms involving proinflammatory cytokines in major depression can cause CHD [71–73]. People with a depressive illness have a dysregulation of immune mechanisms involving pro-inflammatory cytokines, which include elevated levels of interleukins (IL) such as IL-1, IL-2, IL-6, tumour necrosis factor and acute phase proteins such as C-reactive protein [71–77]. Increased levels of these inflammatory markers have been linked to cardiac diseases such as congestive heart failure, CHD, and MI [78–80].
Atherosclerotic lesions have been postulated to stimulate a chronic, low-grade immune activation, resulting in increased production of cytokines [81]. Conversely, inflammation may be linked to an increased rate of atherosclerosis, which in turn confers greater risk of adverse cardiac events [82]. Atherosclerosis is not only a risk factor for CHD, but it has also been suggested that atherosclerosis independently causes both depressive and cardiac illness, possibly simultaneously [83]. Increased levels of cytokines can result in depressive symptoms such as loss of appetite, fatigue, as well as apathy and social withdrawal [83]. Attempts to show that there is a cumulative effect with regard to cytokine levels in patients with depression as well as heart disease have been inconclusive, however [12,84]. If both depression and illnesses such as CHD are caused by a third entity such as atherosclerosis, this may explain such a finding.
In addition, it has been suggested that autonomic neural pathways reflexively monitor and adjust the inflammatory response. A review by Tracey [18] has postulated the existence of a neuro-inflammatory reflex, where inflammatory stimuli activate sensory pathways that relay information to the hypothalamus, which in turn activates an anti-inflammatory response. There is evidence showing that the neural control of acute inflammation is reflexive and can inhibit the activation of macrophages and the release of cytokines via the vagus nerve. Thus, if stimulation of the vagus nerve can inhibit the release of cytokines and counteract an inflammatory response [18], it could thus be postulated that vagal dysfunction could result in increased inflammatory markers, contributing to CHD.
The relationships discussed above are depicted in Figure 3. Empana et al. [80] also found that depressed mood is related to CHD even after adjustment for inflammatory markers, suggesting that they are not the sole mechanisms accounting for the association between depression and CHD.
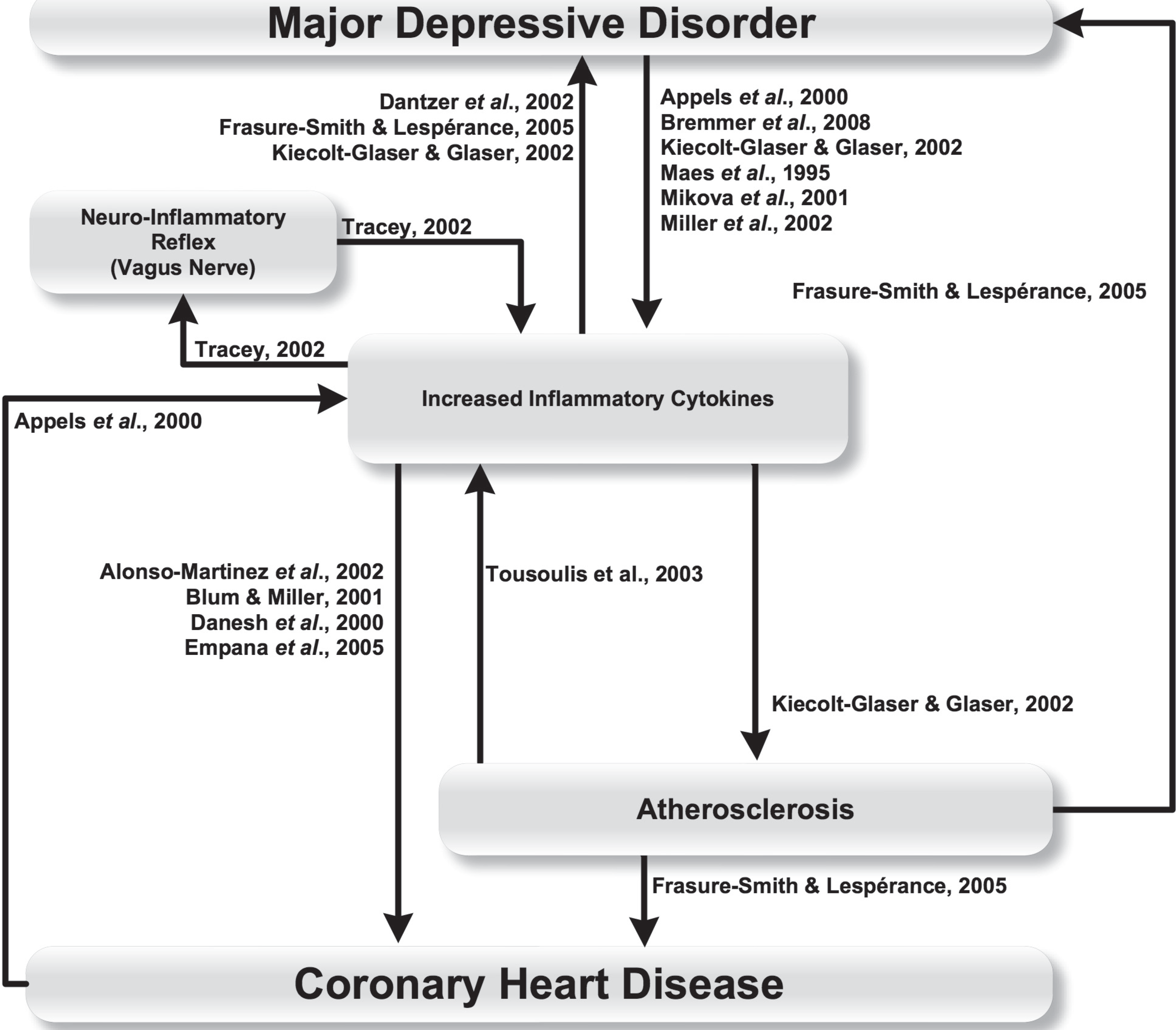
Inflammatory mechanisms linking major depressive disorder and coronary heart disease.
Endothelial dysfunction and platelet activation
Coagulation abnormalities and vascular endothelial dysfunction are thought to cause the development of, or acceleration of, atherosclerosis in people with depression, via raised platelet activation or increased levels of immune cells [85–87]. There is good evidence for a link between mental stress and endothelial function. Acute psychological stress was shown to cause transient endothelial dysfunction in healthy men with no known risk factors for cardiovascular disease [88]. In another study on healthy subjects with no cardiovascular risk factors, a three-minute mental stress task resulted in prolonged endothelial dysfunction [89]. Both studies provide evidence for a link between psychological stress and atherogenesis. Evidence shows that endothelial function also becomes impaired in depressed people [90]. Depression was associated with endothelial dysfunction in a study of 193 post-menopausal women [91]. Although the study finding is expressed in terms of the psychosocial trait of depression/anxiety, depression was measured using the Beck Depression Inventory and correlated with endothelial dysfunction. A similar finding of endothelial dysfunction was made in a study of antidepressant naïve depressed young adults diagnosed using Diagnostic and Statistical Manual IV criteria for MDD [92]. In patients who have suffered an acute coronary syndrome, those who are depressed have higher levels of cellular adhesion molecules such as ICAM-1 [93]. Even in medically healthy patients, depressive illness leads to increased levels of ICAM-1, which predisposes to increased risk of CHD [80].
Depression has also been linked to increased risk of cardiac thrombotic events via platelet reactivity and activation [83]. Upon activation, platelets secrete serotonin, which acts on platelet serotonin receptors to promote aggregation on the platelets [94,95] and causes vasoconstriction of arteries [12,96]. Depressed patients show an up-regulation of serotonin receptors on platelets, leading to increased density of platelet serotonin receptors [97,98] and changes in platelet reactivity and activation [99–101]. Platelet activation can cause increased thrombosis, arterial occlusion and vasoconstriction [7]. Platelet activation likewise increases the levels of inflammatory cytokines [102]. Both endothelial dysfunction and platelet activation in depressed people contribute to atherosclerosis and vascular damage [85–87,102], as well as abnormal brachial artery flow-mediated vasodilation [92]. Atherosclerosis is suggested as compromising cerebral blood supply, leading to damage and neuronal loss in brain regions involved in mood and cognition, resulting in major depression (this concept has been termed the vascular depression hypothesis) [83,103].
These relationships are shown in Figure 4.
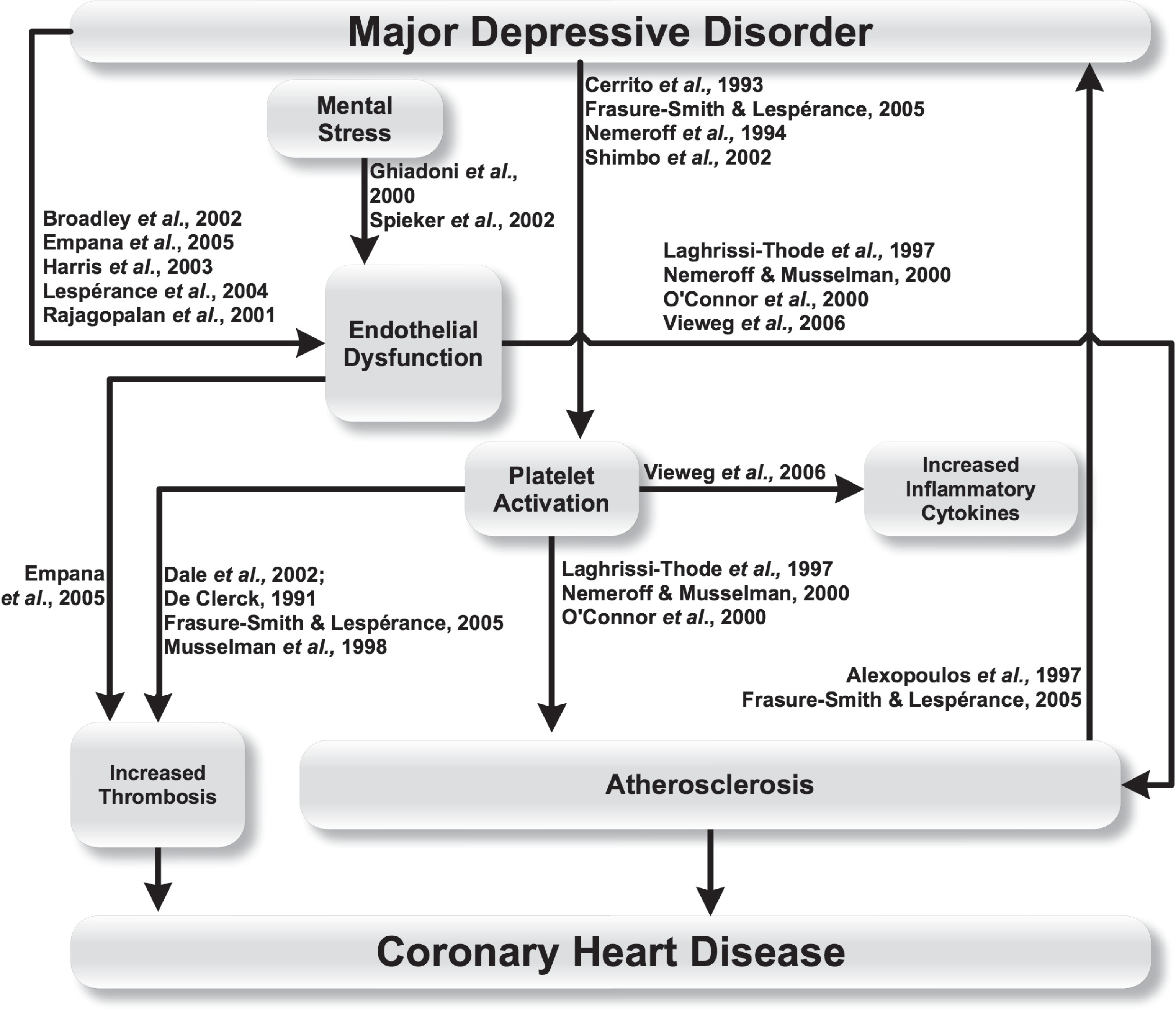
Endothelial and coagulopathic mechanisms linking major depressive disorder and coronary heart disease.
Polyunsaturated omega-3 free fatty acid deficiency
Low serum and low red blood cell levels of omega-3 polyunsaturated fatty acids (PUFAs) are associated with MDD in patients with CHD [104–106]. Omega-3 fatty acid deficiency is also associated with depression in the absence of other medical illness [107–112]. Conversely, eating foods containing PUFAs such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) have been shown to reverse depression and enhance antidepressant efficacy [113–115]. Omega-3 fatty acid deficiency is separately associated with CHD [116]. It has even been proposed that red blood cell EPA plus DHA be considered as an ‘omega-3 index’ to predict risk for death from CHD [117].
PUFAs form constituents of cell membranes [118]. EPA and DHA in particular are found to be concentrated at neuronal synapses and are considered to play an important role in neurotransmission and receptor function in the human brain [116,118,119]. As such, this may represent a direct mechanism by which omega-3 PUFA deficiency causes MDD.
However, it has also been shown that major depression in acute coronary syndrome patients is associated with significantly lower plasma levels of omega 3 PUFAs [104]. Deficiency of these essential fatty acids could thus also represent a mechanism which links MDD and CHD [104]. There is evidence that a dietary deficiency of PUFAs is associated with an increased risk of inflammation [120] and that the effects of hypertensive, atherosclerotic and chronic inflammatory disorders may be exacerbated by inadequate intake of PUFAs [121]. The casual pathways by which PUFAs link CHD and MDD (Figure 5) may thus primarily involve inflammatory mechanisms and atherosclerosis.
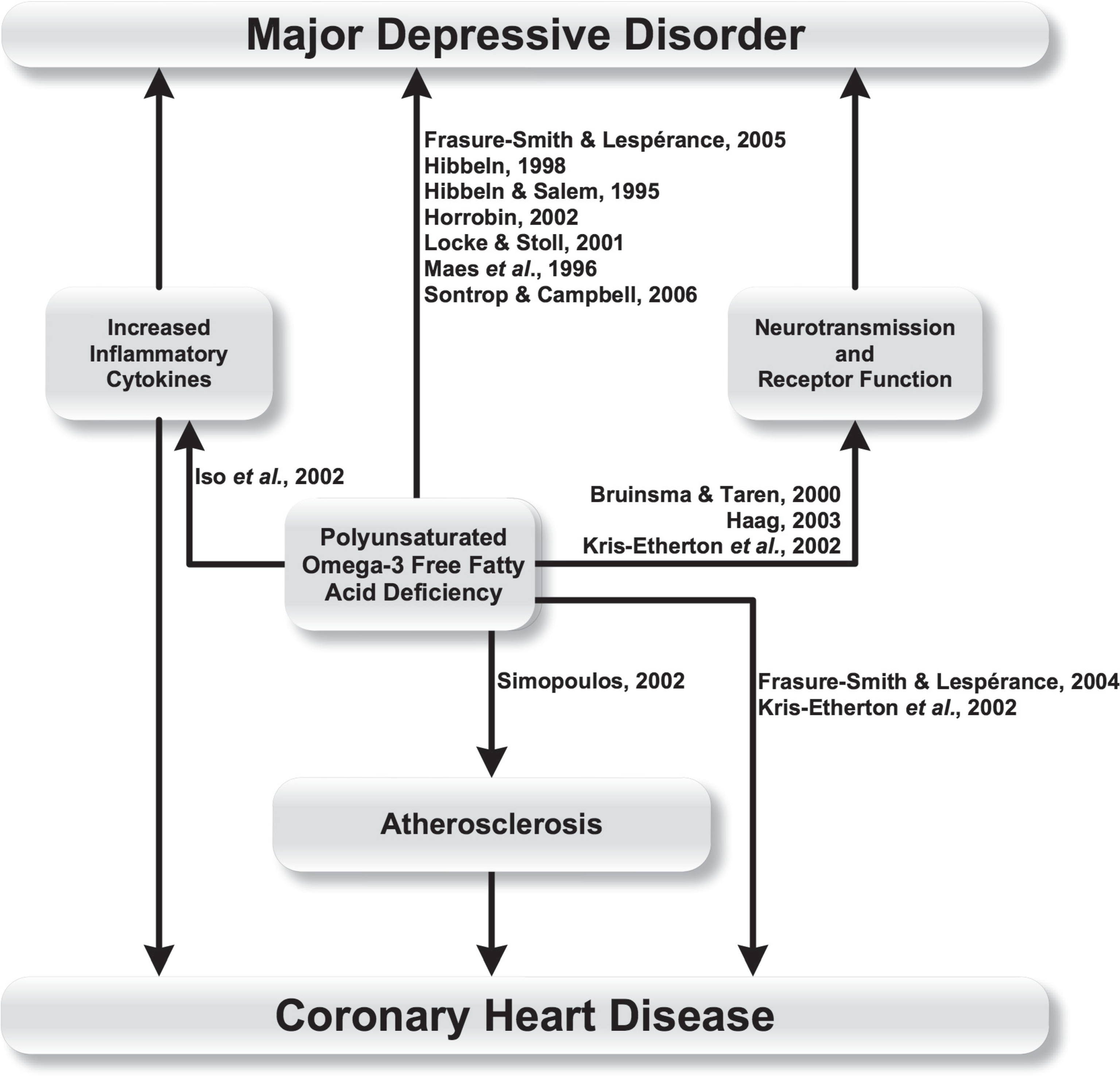
Polyunsaturated omega-3 free fatty acid deficiency linking major depressive disorder and coronary heart disease.
Autonomic mechanisms
Autonomic dysregulation, caused by depression, is thought to result in altered function of the sympathetic nervous system, activation of the hypothalamic-pituitary-adrenal axis, as well as parasympathetic changes, with irregularities of vagal control. This autonomic dysregulation has in turn been linked to an increased risk of cardiovascular disease [7].
People with depression have increased sympathetic activation [5,6], an increased resting heart rate, increased heart rate responses to physical stressors, impaired baroreflex sensitivity, high variability in ventricular repolarization, and reduced heart rate variability [122,123]. These changes in turn have been associated with increased mortality and cardiac morbidity [124]. The causal pathways discussed below are summarised in Figure 6.
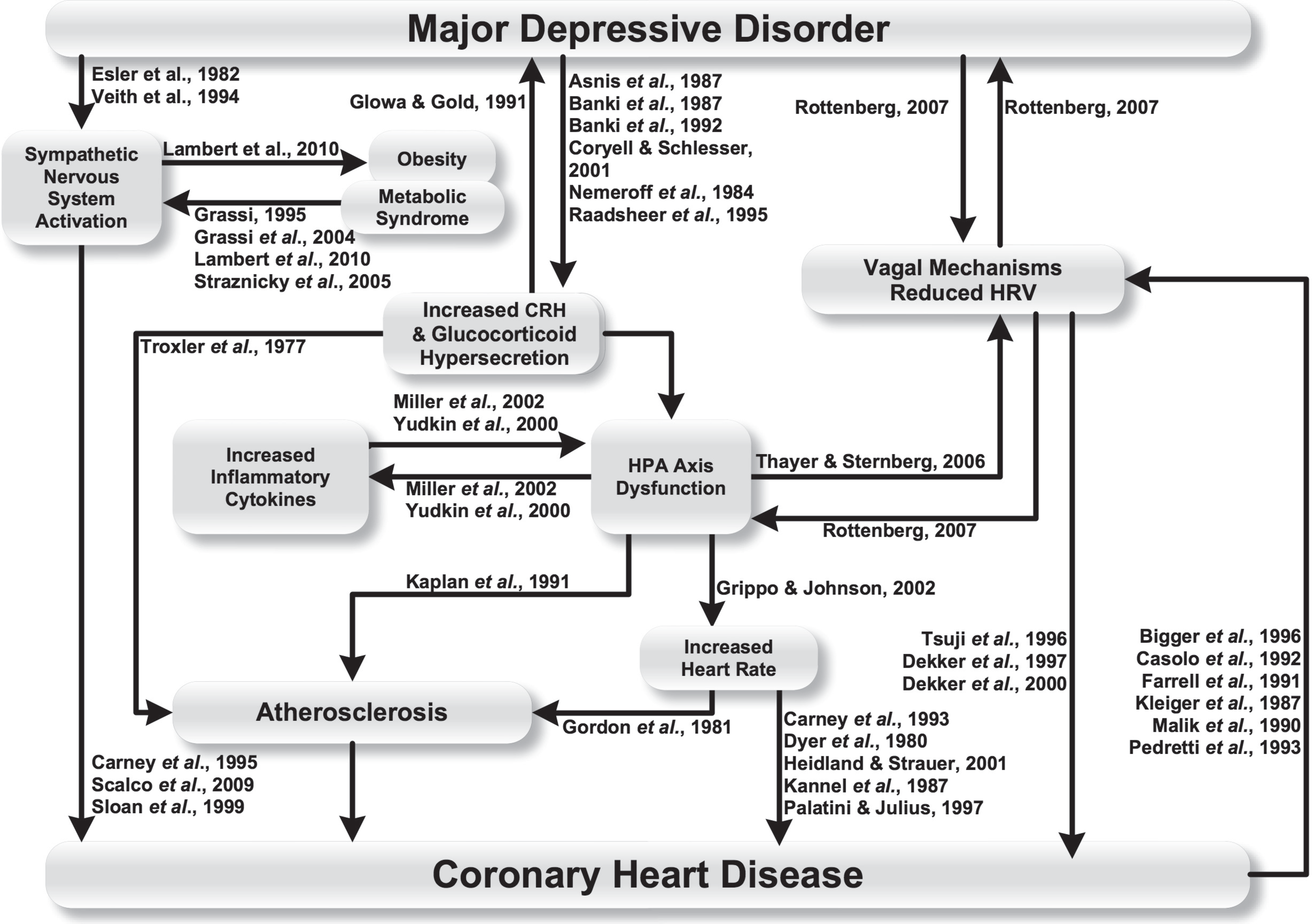
Autonomic mechanisms linking major depressive disorder and coronary heart disease.
Sympathetic nervous system
People suffering from MDD can have increased activation of the sympathetic nervous system [125,126]. Furthermore, sympathetic hyperactivity has been implicated in increased cardiovascular morbidity and mortality in people suffering from MDD [127,128]. Depressed people have been found to have increased levels of sympathetic tone as evidenced by elevated noradrenalin spill-over [125,126]. Activation of the cardiac sympathetic outflow occurs under conditions of experimental mental stress [129] and increased sympathetic outflow has also been associated with the severity of symptoms of depression during experimental mental stress [130–133]. Reaction to stress is generally characterized by an increase in sympathetic nerve activity, which results in physiological changes including increased cardiac output and heart rate, as well as increased muscular blood flow [134]. The increase in these physiological parameters is proposed to be a causal factor between emotional stress and adverse cardiovascular events [134].
Barton and colleagues [135] compared whole body sympathetic outflow with cardiac sympathetic outflow in people with MDD versus healthy controls. The group of participants with MDD as whole did not display elevated sympathetic activity compared with the healthy control group. However, there was a bimodal distribution of cardiac and whole body sympathetic nerve activity in the participants with MDD, with one subgroup who displayed marked sympathetic activation and a subgroup with low sympathetic activity. Such findings allude to the underlying complexity of sympathetic regulation in MDD [135]. Cardiac and total sympathetic activity in patients with MDD has been found to be elevated, while muscle sympathetic activity is unchanged or reduced [135,136]. Such findings support the conclusion that sympathetic nervous activity, at rest and in response to stressors, is regionalized [129]. The sympathetic outflow to organs such as the heart is preferentially activated, particularly during mental stress [129,135] and this in turn is linked with a higher risk of CHD [127,128].
Sympathetic nervous activation has also been shown to occur in obesity and metabolic syndrome [137]. While several mechanisms have been proposed to explain the primary etiology of obesity and obesity-related metabolic disturbances (reviewed in Lambert et al. [138]), sympathetic nervous activity has been shown to be elevated in obese individuals [139,140], and sympathetic outflow changes are considered to be a significant contributor to obesity and its related metabolic sequelae [138]. Obesity has been associated with increased urinary noradrenaline and normetanephrine levels [141,142]. Obese people have also been shown to have increased efferent muscle sympathetic nerve activity [139,140] as well as increased rates of noradrenergic spill-over to the blood from the kidneys [143]. The effects of sympathetic activation in obese people have been linked to cardiovascular pathology such as CHD and cardiac failure via pathways such as hypertension and renal pathology [138].
Hypothalamic–pituitary–adrenal axis
Depressed mood and stress can cause dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis [22]. The HPA axis is the physiological system involved in response to stress [144]. This stress response is also known as the ‘fight or flight’ response [145] or ‘general adaptation syndrome’ [146].
It has been proposed that the general adaptational syndrome is governed by two systems, the corticotropin-releasing hormone (CRH) system and the locus coeruleus-norepinephrine (LC-NE) (sympathetic) nervous system [147–149].
In response to a stressful stimulus, corticotropin-releasing hormone (CRH) is released from hypothalamic neurons, triggering the secretion of adrenocorticotropic hormone (ACTH). ACTH in turn results in cortisol and catecholamine secretion, primarily of adrenaline, but also of noradrenaline. These secretory molecules complete a feedback loop by acting on the pituitary, hippocampus and hypothalamus to modulate further HPA activity. The sympathetic nervous system is also activated [149]. The result is an increase in circulating glucose, heart rate, and blood pressure [149].
The relationship between depression and HPA dysfunction was explored as early as the 1970s, when dexamethasone testing was put forward as a diagnostic test for endogenous depression [150]. In healthy control subjects, dexamethasone administration causes decreased cortisol secretion in response to down-regulation of the HPA axis. However, up to 50% of adult depressed patients have no suppression of cortisol with administration of dexamethasone, due to glucocorticoid hypersecretion [151,152]. Depressed people also have increased levels of corticotropin-releasing hormone (CRH) in the cerebrospinal fluid, as well as in the paraventricular nucleus of the hypothalamus, supporting the hypothesis of an up-regulation of the HPA axis in depression [153–156]. Intra-cerebroventricular administration of CRH in rat [157] and primate models [158] induced decreased levels of feeding and sexual activity, sleep disturbance and learning impairment, behaviours which have been suggested to imply depressed states in these animals.
Increased heart rate has been implicated in the increased risk of adverse cardiovascular events in depression via HPA axis dysregulation [124]. Corticotropin-releasing hormone increases sympathetic activity, which can result in increased mean heart rate [159] in patients with depression [160]. Carney et al. [161] showed that depressed patients with CHD exhibit higher mean heart rates than non-depressed patients with CHD. Increased mean heart rate has been shown to independently be a risk factor for increased coronary artery plaque rupture [162], providing a further mechanism for adverse cardiac events in depressed patients.
The increase in heart rate occurs in otherwise healthy depressed subjects, as well as those with co-morbid cardiac disease [163]. Elevated heart rate has been associated with arrhythmias and sudden death, as well as myocardial ischaemia, and cardiac failure [161,164–166]. In addition, increased heart rate causes increased arterial wall stress and is a risk factor for atherosclerosis [167].
There is also a relationship between elevated plasma cortisol and early atherosclerosis [168]. Chronic HPA and sympathoadrenal hyperactivity have also been linked to atherosclerosis [169]. These links may provide a further mechanism as to how increased plasma cortisol levels increase the risk of CVD. In addition, there is evidence that patients with Cushing's disease, who have increased cortisol levels, have an increased risk of cardiovascular disease [170]. Sympathetic hyperresponsiveness has also been linked to the development of myocardial ischaemia during exercise and mental stress [171]. Krantz et al. [172] also showed that patients with coronary artery disease who had greater cardiovascular reactivity had higher rates of mental stress-induced myocardial ischaemia.
The prefrontal cortex is intimately connected with the limbic system and is assumed to be involved in the modulation of autonomic responses to stress and emotional stimuli. Davydov et al. [122] suggested that the prefrontal cortex is part of the limbic system in terms of modulation of autonomic response. While such a classification might be controversial, there is evidence that the prefrontal cortex directly modulates cardiovascular control [173]. It is thus important in mood regulation and in cortical control of the HPA axis and the sympathetic nervous system. Depressed patients have been shown to have significant loss of cells in the prefrontal cortex [174]. This provides a possible mechanism for autonomic instability or dysfunction in people with depression.
Furthermore, HPA axis dysfunction has been implicated in increasing the levels of pro-inflammatory cytokines [76,175]. There is evidence that the autonomic nervous system may have an effect on the level of pro-inflammatory mediators in the body by modulating certain endocrine functions [176]. Adrenergic modulation by the sympathetic system and also the HPA axis can result in either the increased or decreased release of pro-inflammatory cytokines such as tumour necrosis factor (TNF) and interleukin 1 (IL-1). Inflammatory cytokines, such as IL-6, may in turn further stimulate the HPA axis, which leads to the increased secretion of cortisol and other hormones [76,175]. In addition, HPA axis dysfunction and reduced cardiac vagal control can result in reduced heart rate variability (HRV) [177].
Parasympathetic nervous system: vagal mechanisms and heart rate variability
In humans, heart rate is not constant. It varies considerably, subject to the cardiovascular demands placed on it and the varied control mechanisms acting in a complex network of feedback loops [127,178]. HRV measures the beat-to-beat variations in heart rate, which naturally occurs in normal cardiac function. HRV is a key measure of the autonomic regulation of cardiac function and ‘healthy’ or normal cardiac function is determined by these feedback loops acting on the heart [127,179]. The small delays in each feedback loop cumulatively result in complex oscillations in heart rate which are measurable by beat-by-beat time series [169]. This produces a complex non-linear variation of heart rate. HRV has been linked to cardiovascular health outcomes [178]. It has been suggested that poor health is associated with a loss of complexity and a correlation exists between severity of illness or dysfunction and magnitude of loss of complexity in cardiac control. Depression and CHD are theorized to disrupt autonomic control feedback loops causing a reduction in HRV [127]. HRV is thus a key method for exploring directly the vagal (parasympathetic) influence of the brain on the heart, and vice versa, which may provide a strong and direct neurological mechanism linking depression and CHD.
Parasympathetic cardiac control is the principal mechanism for autonomic regulation of cardiac function [180] and is mediated by the vagus nerve [181]. Changes in vagal tone have been linked to both depression and cardiac disease [180]. The control of physiological cardiac parameters such as heart rate are hypothesized as being the result of a dynamic equilibrium between opposing sympathetic and parasympathetic (vagal) influences [182]. This dynamic equilibrium, which also results in HRV, is thought to occur due to co-activation or co-inhibition of the sympathetic and parasympathetic nervous systems, or an activation of one with an inhibition of the other [182]. However, vagal tone is thought to provide overall control of the equilibrium by acting as a ‘brake’, counteracting and damping sympathetic drive, resulting in the term cardiac vagal control (CVC) to refer to the balance of both sympathetic and parasympathetic control elements [180]. The level of resting vagal tone is linked to physiological responses to stressful situations. High baseline levels of CVC allows appropriate regulation of sympathetic and parasympathetic balance. Weak parasympathetic control in turn may be linked to poor balance of parasympathetic and sympathetic control [180].
Depression can cause reduced CVC [180]. Reduced HRV may in itself present a further mechanism relating depression and CHD, as HRV is a known risk factor for sudden death and ventricular arrhythmias in patients with CHD [183–185]. Early HRV changes have been shown to occur following myocardial infarction (MI) and have been linked prognostically to mortality risk [186–191]. The relative risk of mortality after acute myocardial infarction is significantly higher in patients with decreased HRV [188,192]. A decline in HRV increases the risk of myocardial infarction and coronary insufficiency [185], increases the relative risk of death from cardiovascular disease [193,194] and increases the risk for sudden cardiac death [195,196]. It should be noted, however, that low HRV has also been associated with increased risk of death in individuals without hypertension, diabetes, cancer or symptomatic heart disease [193].
Finally, it has been suggested that there may be evidence for a causal relationship between CVC and depression and that CVC may be involved in depression maintenance [180].
Relationship between major depression and coronary heart disease: a ‘causal network’.
From the review of the literature, it is evident that there are several possible mechanisms responsible for the observed relationship between depression and cardiac disease. They are often presented as separate entities in the literature [17,20]. However, it is conceivable that no one mechanism stands alone as a cause for the relationship. In fact, the causal mechanisms described in this review are linked and function in concert, rather than a group of isolated pathways linking depression and ischaemic cardiac disease. There are thus considerable inter-relationships between mechanisms and even the possibility of feedback loops between pathways. This suggests that the mechanisms might form a ‘causal network’, which describes the relationship between MDD and CHD. This causal network thus describes all the linked mechanisms connecting MDD and CHD, conceptualizing them as a single interconnected system. A diagram of the proposed causal network, based on current evidence, is shown in Figure 7.
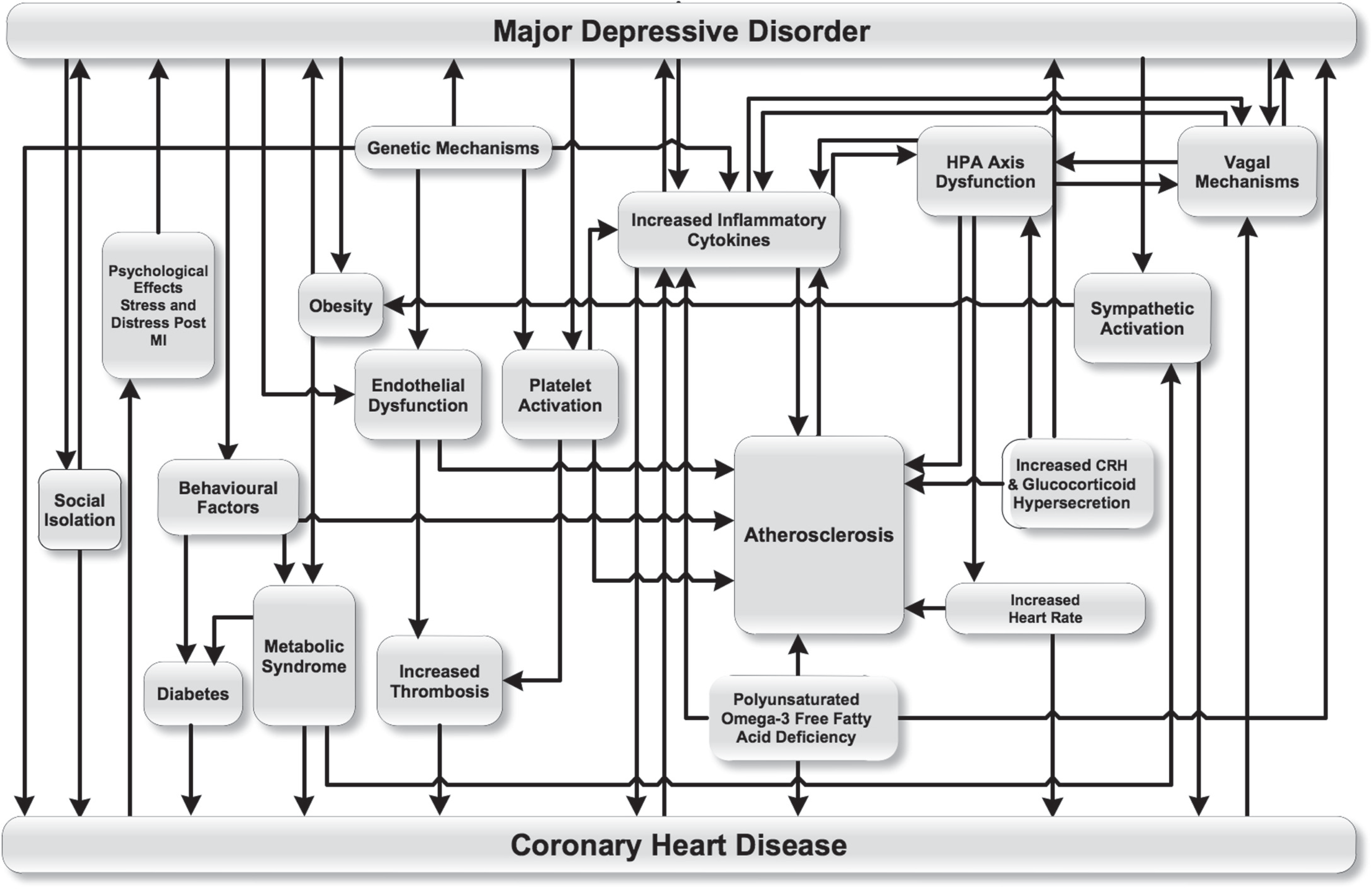
The relationship between major depressive disorder and coronary heart disease: a topographical map of the causal network.
Barth et al. [17] describe cytokine, endothelial and autonomic nervous system mediated pathways as ‘direct’ pathways between MDD and CHD. The authors then describe behavioural mechanisms, such as the increase in classic coronary risk factors with depression leading to CHD, as ‘indirect’ pathways. However the overall synergistic effect of multiple interrelated pathways linking cardiac disease and depression may mean that no single pathway is solely ‘direct’. Additionally, behavioural factors such as smoking may feed directly into the causal network by accelerating processes such as atherosclerosis [197], further blurring what is considered a ‘direct’ and ‘indirect’ pathway.
Having described how MDD and CHD are linked by a network of causal pathways, how does this concept further promote understanding of the complex interrelationship between these two disease entities? The causal network is best conceptualized as a biological network. As such, it is amenable to further study within the framework of systems biology [19]. In order to establish how systems biology can be applied to the causal network, a brief introduction to systems biology itself is required.
Discussion
Application of network theory to the causal network linking major depression and coronary heart disease
Systems biology, which encompasses network theory and its methodological approaches, has shown great promise, offering new tools to analyse and understand complex biological systems. Systems biology involves an integrated analysis of interacting biological pathways or networks [198]. Large numbers of individual elements (such as bio-molecules) or even biological sub-systems, are measured simultaneously over time and their relationships to each other are elucidated. Complex networks occur widely, with common examples including societal and social networks [199,200], co-authorship of scientific papers [201,202], and the internet [203,204]. Biological systems in particular are amenable to elucidation using network theory, with examples ranging from networks of biochemical interactions or gene networks [199,205], protein and enzymatic networks [199,206], through to metabolic pathways [205] and food webs in ecology [207,208]. The principles of biological networks also apply to disease mechanisms. Noorbakhsh et al. [198] have argued that systems biology methodology has immediate application in understanding complex diseases which involve multiple pathogenic determinants. The authors make this point in the context of neurodegenerative diseases such as Alzheimer's disease [198]. It is equally applicable to the multiple and complex pathogenic determinants linking CHD and major depression. It is thus proposed that the causal network underlying CHD and major depression is amenable to analysis using the paradigm of systems biology.
There have been proposals to map, understand, and model, in quantifiable terms, the topological and dynamic properties of various biological networks, for example those that control the behaviour of living cells [209]. Alm and Arkin [199] have suggested that network topology data can be used to generate new hypotheses about how systems are organized. By further studying how the various pathways which link CHD to depression are interconnected with each other, understanding could be reached on how to reduce the risk of recurrent cardiac events in depressed CHD patients, for example.
Establishing the topology of the network
By conceptualizing the causal network as a biological network, it follows that the topology of this network needs to be ascertained. Figure 7 represents an initial attempt to provide a topological map of the causal pathways linking MDD and CHD. Applying the paradigm of systems biology would allow the use of several tools to establish in detail the topology or overall structural organization of the causal network. Specific measures such as network degree, degree distribution and the clustering coefficient [209] could be used to characterize the causal network.
Future work should assess whether the causal network has characteristics of a modular network (in which each node should have about the same number of links), a scale-free network (in which most pathways are linked through a few highly connected ‘hubs’) or a hierarchical network, which has characteristics of both scale-free and modular networks [199,210]. If, for example, the causal network has properties of a scale-free network, it means that certain nodes are highly connected and thus critical to the causal relationship between MDD and CHD. For instance, inflammatory cytokines (see Figure 7) might represent such a highly connected hub in the relationship between CHD and MDD. This might mean that by focusing clinical prevention or treatment on this mechanism, such treatment or prevention may be more effective.
Quantification of the causal network
The emergent properties of a biological system (or network) generally cannot be inferred from the characteristics of isolated components or pathways of that system. Systems biology relies on large-scale quantification of biological systems, with subsequent integration of the quantified data, which allows the development of accurate models of the system [198]. Such models provide information not only on the architecture of a given network, but also on the dynamics of the network (i.e. how information, energy or other elements flow through a given network). Each mechanism linking major depression and CHD can be conceptualized as a node in a causal network. Attempting to quantify each node will lead to an understanding of which nodes contribute more, or less (in quantitative terms), to exacerbating cardiac disease in depressed patients, or causing depression in patients post MI. Despite being interrelated, it is not clear how much each mechanism may contribute to the overall relationship between MDD and CVD in quantitative terms. If quantitative relationships can be established between each pathway, and between measures of depression and cardiac outcomes, potentially the contribution of each mechanism or pathway can be measured and ranked.
Conclusions and recommendations
This review has described several mechanisms which have been put forward as causes for the link between major depression and CHD. Behavioural mechanisms, genetic mechanisms, dysregulation of immune mechanisms, coagulation, omega-3 PUFA deficiency, as well as vascular endothelial abnormalities and autonomic mechanisms, have previously been discussed as separate entities in the literature. However, the considerable overlap and inter-connectivity between them suggests that they form a causal network which links MDD and CHD. Viewing these causal mechanisms as an interdependent network, which is amenable to analysis using a systems biology approach, presents a new paradigm in this field and provides fertile ground for further research. The initial network model presented in this review is incomplete and presents a starting point. Further research is required to establish all the nodes and links in the network (i.e. further mechanisms which may link CHD and depressive illness, as well as further links between them). However, it is hoped the following recommendations will guide further research in this field:
More research is needed to further the complete characterization of the causal network linking MDD and CHD, allowing identification of all the interlinked mechanisms connecting the two diseases.
The establishment of the topology of the causal network will allow the most accurate description (and subsequent modelling) of how all the mechanisms relate to each other in terms of network theory.
The quantification of activity between each node of the network, along with a model of the network topology, will allow the identification of the most significant nodes in the causal network.
Since these significant nodes will correspond to a specific metabolic or disease process (e.g. the increased release of inflammatory cytokines), these nodes or processes can be specifically targeted to yield an efficient means of disease prevention or treatment. It is likely that if a key node in the network is targeted effectively, this intervention may have a significant effect on both MDD and cardiac risk in patients.
Adopting a systems biology paradigm would allow analysis using techniques which already exist, to qualitatively and quantitatively describe this causal network and would provide fertile ground for further research.
Footnotes
Acknowledgements