
Abstract
Schizophrenia includes positive, negative, and cognitive symptoms. Negative and cognitive symptoms do not benefit from current treatments and currently are the main determinants of functional outcome. In the European Union, where healthcare is widely accessible, 80%-90% of patients with schizophrenia are unemployed, while 10% of them die by suicide. Currently, it is believed that psychosis and schizophrenia’s positive symptoms stem from excessive dopamine D2 activity in the striatum, leading to ‘hyper-salience’ followed by delusions, and in the sensory cortex leading to ‘self-generated sensory activity’ followed by hallucinations. The reviewed evidence in this article suggests open potassium and hyperpolarization-activated cyclic nucleotide-gated (HCN) channels leading to prefrontal cortex (PFC) dysfunction followed by ‘cognitive impairment’/’loss of insight’/’lack of deliberate reasoning’/’lack of reality monitoring’ to also be a contributing factor in psychosis and schizophrenia. This could explain how kappa-opioid agonists and potassium channel openers induce psychosis while lowering dopamine but opening potassium channels; how nicotine improves certain schizophrenia symptoms while increasing dopamine but closing potassium/HCN channels; how insightfulness is maintained with 5HT2A psychedelics which increase dopamine but close potassium channels; why guanfacine which closes potassium/HCN channels is the best treatment in delirium psychosis which is characterized by prominent cognitive dysfunction; and why clozapine which closes potassium/HCN channels is superior to other antipsychotics. This article concludes that having a cognitive deficit in the first place may make someone more susceptible to developing all schizophrenia symptoms and that potassium/HCN channel blockers would improve that. They would especially ameliorate the neglected cognitive and negative symptoms. This article also notes the importance of norepinephrine and NMDA. Lastly, it proposes treatment perspectives, summarizes the reviewed findings in Table 1, and presents theorized pathways behind schizophrenia and psychosis in Figure 1.
1 Introduction
Schizophrenia is a severe mental disorder affecting 1% of the population [1]. It includes positive, negative, and cognitive symptoms. At present, schizophrenia’s treatment consists of antipsychotics. Current antipsychotics are based on the theory that excessive dopamine D2 activity leads to psychosis and that high 5HT2A activity and low NMDA activity can contribute to those high dopamine levels [2]. Those antipsychotics work well against positive symptoms, still certain patients’ positive symptoms do not respond to treatment [1]. Those treatment-resistant patients may benefit from a treatment with a different mechanism of action such as modulating ion channels. In addition, negative and cognitive symptoms do not benefit from current treatments and currently are the main determinants of functional outcome [3-6]. In the European Union, where healthcare is widely accessible, 80%-90% of patients with schizophrenia are unemployed [7], while 10% of them die by suicide [8]. Some patients even present schizophrenia with mainly negative symptoms and thus current treatments are of little benefit to them [3, 4]. Modulating ion channels may bring improvements to the overlooked symptoms of schizophrenia, namely the cognitive and negative symptoms. Current treatments often also come with various side effects, such as cardiotoxicity [9], increased type 2 diabetes risk [10], hyperprolactinemia [11], and extrapyramidal side effects [12]. Antipsychotics can also lower reward sensitivity and thus motivation [13]. Second-generation antipsychotics using 5HT2A as a target have fewer side effects. Still, insidiously, 5HT2A antagonists could increase submissive behavior at the detriment of leadership, social function, and attraction of males towards females [14, 15]. Modulating ion channels may come with fewer side effects. To conclude, schizophrenia treatments have a need for improvement; as this article contemplates a new treatment perspective it is of clinical relevance. In addition, the article is of clinical relevance due to having implications for current research on potassium channel modulators and the NMDA activating drug iclepertin [16, 17]. It even contraindicates ongoing research on potassium channel openers in the treatment of schizophrenia [18, 19].
The theory that will be contemplated in this article stemmed from the knowledge that certain forms of psychosis come with severe cognitive impairment during the psychotic break, leading to a loss of insight. This cognitive impairment could come from an ‘offline’ PFC. Indeed, cognitive dysfunction precedes patient’s first psychotic episode [20], poor insight is associated with executive and PFC dysfunction [21, 22], reduced activity in the medial PFC is associated with reduced reality monitoring that may lead to misattribution of ‘self-generated sensory activities’ as hallucinations [23], and individuals with psychosis seem to not deliberately engage in reasoning which may lead to delusions [24]. As a result, this article theorizes that prefrontal dysfunction could contribute to psychosis. This would indicate that multiple factors can contribute to psychosis, namely, ‘hyper-salience’ leading to delusions from striatum hyper-activation [25, 26], ‘self-generated sensory activity’ leading to hallucinations from hyper-activation of the sensory cortex [23], and, as theorized in this article, cognitive impairment from an ‘offline’ PFC. The ‘offline’ PFC would
limit reasoning ability, while the simultaneous hyper-activation of the striatum would create an emotional bias of ‘hyper-salience’ [27], leading to delusions. Also, an ‘offline’ PFC would limit reality monitoring ability, while hyper-activation of the sensory cortex would increase ‘self-generated sensory activities’, leading to hallucinations. As a result, this article will investigate the ‘cognitive impairment’/’loss of insight’/’lack of deliberate reasoning’/’lack of reality monitoring’ factor with the premise that this factor, together with ‘hyper-salience’ or ‘self-generated sensory activity’ will be more or less present in different forms of psychosis. Because potassium channel closure is important in keeping the PFC ‘online’ [28], this article will look at how potassium channels relate to pathways and treatments known to affect psychosis. In addition, the current literature on potassium channels in schizophrenia and each of its symptoms will be reviewed. Similarly, HCN channels will also be investigated as those too when opened can contribute to an ‘offline’ PFC [29]. To conclude, this article’s final theory is that ‘cognitive impairment’ due to the PFC being ‘ offline’ during a psychotic break contributes to ‘loss of insight’/’lack of deliberate reasoning’/’lack of reality monitoring’ and psychosis itself and that excessive potassium/HCN channel opening is largely involved in this.
2 Norepinephrine on ion channels
As the following findings suggest, norepinephrine is relevant because it could contribute to psychosis. For example, it has been hypothesized that high norepinephrine can contribute to psychosis, especially of the paranoid subtype, in certain schizophrenia patients: with some evidence behind it [30, 31]. Current antipsychotics primarily act on dopamine and less on norepinephrine which could explain why paranoid subtypes are more treatment-resistant [1]. Impaired sensory gating is found in schizophrenia and psychosis, which leads to attentional impairment, and is used as a psychosis indicator [32]. Opioids lower the release of norepinephrine while they improve sensory gating and can provide an antipsychotic effect [33-36]. Clonidine, which selectively lowers norepinephrine, also improves sensory gating and psychosis [37-40]. Increased prefrontal norepinephrine contributes to salience attribution and thus could contribute to the ‘hyper-salience’ aspect of schizophrenia [41], leading to delusions. High norepinephrine acts on a1-adrenoceptors (a1-AR) and can take the PFC ‘offline’ [28, 42]. To conclude, norepinephrine could contribute to psychosis via ‘hyper-salience’ and PFC impairment.
The PFC impairment could be due to norepinephrine modulating ion channels. Moderate norepinephrine levels act on a2-adrenoceptors (a2-AR) which decreases cAMP-PKA opening of potassium channels [28, 42]. Norepinephrine a2-AR also lowers HCN channel opening via decreased cAMP-PKA [43-46]. On the other hand, high norepinephrine engages low-affinity receptors such as a1-AR that drive caldum-cAMP opening of potassium and HCN channels, rapidly taking the PFC ‘offline’ [29, 38, 44]. Thus, a2-AR, most active when norepinephrine levels are moderate, lowers cAMP, potassium channel, and HCN channel opening, leading to increased neuronal excitability. On the other hand, a1-AR, most active when norepinephrine levels are high, increases CAMP, potassium, and HCN channel opening, which contributes to taking the PFC ‘offline’. Low norepinephrine would not activate a2-AR and, consequently, not close potassium/ HCN channels, thus leaving them open and the PFC ‘offline’. To conclude, both too high or too low norepinephrine leads to PFC impairment potentially contributing to psychosis, but only high norepinephrine leads to ‘hyper-salience’ contributing to psychosis. High norepinephrine contributing to psychosis while opening potassium/HCN channels to create prefrontal impairment is aligned with this article’s contemplated theory.
3 Dopamine on ion channels
Dopamine is highly relevant to psychosis and schizophrenia. Excess dopamine D2 activity is well known to cause psychosis, while dopamine D1 is not [2]. Downstream effects on ion channels could be of interest. Moderate dopamine mainly acts on D1 receptors and subsequently deactivates inwardly rectifying potassium (Kir) channels among other potassium channels [47-49]. This may explain how moderate dopamine leads to ‘information maintenance’ [50], which refers to persistent maintenance of information in working memory. High dopamine acts on D2 receptors and subsequently activates Kir among other potassium channels [49, 51-53]. This may explain how high dopamine leads to ‘information updating’ [50], which refers to flexible manipulation of information in working memory or set shifting. Additionally, dopamine D1 deactivates sodium channels and opens HCN channels [54, 55]. D1 closing potassium and opening HCN channels leads to membrane depolarization, while closing sodium channels leads to membrane hyperpolarization. A trend can be found with D2 receptors hyperpolarizing membranes. Next to opening potassium channels, they also block sodium channels [56]. Thus, D2 receptors let positive potassium ions flow out of the membrane while not letting new positive sodium ions in. D2 does not let positive potassium ions in either by also blocking HCN channels [57]. To conclude, both high dopamine and norepinephrine can induce psychosis, and both have similar downstream effects on potassium channels. Namely, high or low levels of those neurotransmitters open potassium channels and take the PFC ‘offline’. Thus dopamine’s downstream effects on ion channels is aligned with the theory of open potassium channels contributing to psychosis.
4 Glutamate on ion channels
Glutamate is known to be increased in schizophrenia, while NMDA receptor activity is known to be decreased [2]. Downstream effects on ion channels could be of interest. High glutamate opens potassium channels [58], while moderate levels of glutamate mainly act on metabotropic glutamate receptor 3 (mGluR3) to lower potassium channel activity [28]. In theory, low NMDA receptor activity leads to high glutamate and dopamine levels [2], which, downstream, would open potassium channels. However, the relationship between NMDA receptors and potassium channels is more complex. Homeostatic intrinsic plasticity encompasses the mechanisms by which neurons stabilize their excitability. NMDA and potassium channels act interchangeably to maintain homeostatic intrinsic plasticity and thus maintain neuronal excitability within physiologic boundaries [59]. NMDA receptor activation increases neuronal excitability via calcium influx and subsequently increases Kir channels to normalize excitability back down [60]. NMDA receptor blockade decreases neuronal excitability and subsequently lowers potassium channels to normalize excitability back up [59]. This means that blocking potassium channels could be one way of restoring the lack of neuronal excitability induced by low NMDA activity.
To conclude, similar to dopamine and norepinephrine, high glutamate is associated with psychosis and potassium channel opening. Moderate glutamate levels can close potassium channels and thus low glutamate would again lead to open potassium channels. In addition, low NMDA activity is associated with psychosis, at least in part via a downstream dopamine increase [2]. However, low NMDA activity could also reduce neuronal excitability and contribute to PFC dysfunction. Indeed, the NMDA-increasing drug iclepertin has been shown to improve cognition in schizophrenia [17]. Closing potassium channels could remediate low NMDA activity associated lack of neuronal excitability and thus PFC dysfunction. Indeed, in an animal model of schizophrenia induced by NMDA receptor blockade, the closure of potassium channels was able to normalize neuronal excitation in the PFC, thereby improving cognition [61].
5 Acetylcholine on ion channels
Moderate activity on muscarinic acetylcholine receptor M1 (M1) deactivates KCNQ potassium channels, while high M1 activity activates KCNQ2 potassium channels via increases in cAMP to prevent excitotoxicity and seizures [62]. Thus, similar to dopamine, norepinephrine, and glutamate, moderate activation of M1 could lower potassium channel opening, while high or low activation could increase potassium channel opening. Interestingly, postmortem studies of brains from patients with schizophrenia have reported reduced M1 protein and M1 mRNA levels in the PFC [62]. This makes acetylcholine align with the theory of open potassium channels contributing to psychosis, similarly to previously reviewed neurotransmitters.
6 5HT2A on ion channels
Excess 5HT2A activity is implicated in psychosis [2], and second-generation antipsychotics act in part by antagonizing this receptor [63]. Downstream effects on ion channels could be of interest. 5HT2A activation closes voltage-gated potassium (Kv) channels Kv1.1, Kv1.2, Kv7 [64-66], and possibly others while it also increases the risk of psychosis: which at first sight is not consistent with the theory that potassium channel closure improves psychosis. However, if potassium channel opening only is a factor among others contributing to psychosis, then closing potassium channels may not exclude a certain form of psychosis to occur in a context where striatum hyper-activation or sensory cortex hyper-activation surpasses a certain threshold. In addition, this potassium channel closure could be explained by being a compensation the 5HT2A receptor tries to make before increasing the risk of psychosis or cognitive impairment via increased prefrontal glutamate [67], increased striatal dopamine [68], and decreased prefrontal dopamine/norepinephrine [69], which all are mechanisms that can induce psychosis or open potassium channels as explained above. Thus, after a certain activation threshold, 5HT2A’s direct potassium channel blockade may become surpassed by 5HT2A’s downstream effects on glutamate, dopamine, and norepinephrine, which can open potassium channels and induce psychosis.
Further evidence indicates potassium channel closure via 5HT2A activity to positively impact psychosis via increased insight and cognition as expected when the PFC is not impaired.
An argument for the importance of cognition and insight in psychosis is delirium. In delirium, as cognitive symptoms worsen, the risk of psychosis increases [70]. This article theorizes delirium psychosis to be a form of psychosis with high cognitive impairment as a major contributor. It is interesting that delirium psychosis, a form of psychosis with high cognitive impairment and lack of insight, responds poorly to conventional antipsychotics but does respond well to guanfacine [71]; which mainly acts by closing potassium and HCN channels via increased a2-AR activity and decreased a1-AR activity [45], while even increasing dopamine [46, 72]. This is indicative of an association between psychosis, insight, cognition and potassium/HCN channels.
If insight and cognition are of importance in psychosis it is interesting that 5HT2A affects those positively while increasing psychosis but closing potassium channels. For example, 5HT2A induced psychosis is the only psychosis pathway that comes with retained insight up to a certain point [2]. This may be because 5HT2A is the only psychosis pathway that blocks potassium channels. Additionally, Parkinson’s disease psychosis is associated with better insight during the psychotic break and can be treated with 5HT2A antagonists only, while other psychoses need additional D2 blockade [2]. This is again indicative of 5HT2A and its downstream effects on potassium channels to be associated with insight during the psychotic break, with insight being related to cognition [21, 22]. As last evidence, sensory gating, which is related to cognition in psychosis, is affected ambiguously by 5HT2A agonism which is consistent with 5HT2A activation leading to ambiguous effects on psychosis due to potassium channel blockade [73].
To conclude, the previous information appears to be consistent with potassium channel closure positively affecting ‘cognitive impairment’/’loss of insight’/’lack of deliberate reasoning’/’lack of reality monitoring’ and being a factor more or less active in different forms of psychosis. But it appears that potassium channel closure does not prevent certain forms of psychosis when other contributing factors surpass a certain threshold.
In addition, it is interesting to note that Kv7 channel blockade in the PFC has been shown to suppress 5HT2A activation [66]. This means that selectively blocking Kv7 could improve the PFC impairment aspect of psychosis/schizophrenia, all the while lowering the striatum hyper-activation and sensory cortex hyper-activation aspects via lowered 5HT2A activity.
7 Kappa-opioid on ion channels
Kappa-opioid agonists acutely lower dopamine and still induce psychosis [74-79]. Blocking those receptors would improve psychosis while increasing dopamine [74-76]. This is not consistent with the theory that excessive dopamine D2 activity is responsible for psychosis. However, it is consistent with the potassium channel opening theory as kappa-opioid agonism opens Kir potassium channels in a direct way [80, 81]. Additionally, by lowering dopamine, glutamate [82], and acetylcholine [83], kappa-opioid would further open potassium channels in an indirect way via low D1, M1, and mGluR3 activity. Kappa-opioid agonists negatively affect cognition and insightfulness while opening potassium channels which is consistent with the theory that potassium channel opening negatively affects cognition and insightfulness [75, 79, 84].
The same idea as in the 5HT2A section can be applied here. If excessive dopamine D2 activity leading to striatum hyper-activation or sensory cortex hyper-activation is only one factor contributing to psychosis among others, then lower D2 activity may not exclude a form of psychosis to occur in a context where potassium channel opening surpasses a certain threshold. Confirming this claim, 5HT2A activation and kappa-opioid activation can both induce psychosis while having opposite effects on dopamine, glutamate, and potassium channels. This is consistent with the theory that multiple factors can contribute to psychosis and thus each be more or less present in different forms of psychosis.
In addition, kappa-opioid is implicated in schizophrenia’s negative symptoms. Potassium channels regulate presynaptic D2 receptor function to inhibit dopamine release when activated [53]. Anhedonia is defined as a diminished capacity to experience pleasant emotions and is one of the cardinal negative symptoms in patients with schizophrenia. Kappa-opioid agonists worsen anhedonia and thus negative symptoms of schizophrenia by lowering dopamine [74, 75], as would be expected if they activate potassium channels to inhibit dopamine release.
To conclude, findings on kappa-opioid activity indicates potassium channel closure has the potential to improve psychosis and cognition by preventing PFC impairment, in addition to improving schizophrenia negative symptoms. This suggests potassium channel closure has the potential to improve all symptoms of schizophrenia.
8 Neurodegeneration and ion channels
Neuroinflammation and oxidative stress are relevant because they can contribute to the positive, negative, and cognitive symptoms of schizophrenia in a direct way and together with low brain-derived neurotrophic factor (BDNF) in an indirect way by contributing to the neurodegenerative aspect of the disease [85-92]. Closing Kir4.1 channels facilitates BDNF expression to restore neurodegeneration [93]. Closing the calcium-activated potassium (KCa) channel KCa3.1 and potassium channel Kv1.3 decreases neuroinflammation [94, 95]. Closing Kv channels reduces oxidative stress [96], which is particularly interesting as D2 blocking antipsychotics can increase oxidative stress [97, 98], and cognitive impairment has been shown to be related to oxidative stress in patients with schizophrenia [99]. Thus, closing various potassium channels could improve the neurodegenerative aspect of schizophrenia. Indeed, excessive potassium channel opening contributes to dendritic atrophy, working memory deficits, and explains the amnesia and neurodegeneration caused by cerebral ischemia [29, 100].
Not only potassium, but HCN channels can also impact neurodegeneration. Excessive activation of HCN channels due to chronic stress can lead to prefrontal cortex loss of dendritic spines, and these structural changes are associated with PFC dysfunction and loss of working memory in schizophrenia [101]. Blocking HCN channels could thus prevent these neurodegenerative changes in the PFC. Additionally, HCN2 blockade decreases inflammation in the central nervous system [102].
Inflammation closes potassium and HCN channels [103, 104]. However, opening potassium and HCN channels is a mechanism used to increase neuroinflammation [95, 102, 105], while Kv1.3 and HCN2 channels upregulate after chronic inflammation [102, 106, 107]. Thus, inflammation may partly be associated with the worsening of schizophrenia due to it initially being associated with opened potassium and HCN channels. Also, oxidative stress can result in ion channel malfunction [108], and oxidative stress increases potassium channels [109, 110], which may partly explain how oxidative stress contributes to schizophrenia symptoms.
As previously theorized, psychedelic therapy currently used for major depression could reverse the neurodegeneration found in schizophrenia [86], including the loss of PFC dendritic spines [111]. This neurodegeneration reversal could be necessary for ion channel modulators to provide their full therapeutic effect. However, HCN and potassium channel blockers could be sufficient on their own in restoring neurodegeneration over time by preventing further neurodegeneration while increasing BDNF. Indeed, HCN1 channel blockade is in part responsible for the synaptogenesis and other antidepressant effects provided by (S)-ketamine [112]. As the prevalence of major depressive disorder is high among patients with schizophrenia [113], high HCN1 activity may be an overlapping risk factor for both disorders.
On the other hand, opening potassium channels can create relief from excitotoxicity while excitotoxicity may contribute to the disorder [114, 115]. However, as schizophrenia contains low NMDA activity, this theory seems paradoxical. Also, closing potassium channels lowers neuroinflammation, which is one way of lowering excitotoxicity [116]. Still, high doses of potassium channel blockers should probably be avoided as they could induce excitotoxicity. Alternatively, HCN channels affect excitotoxicity and epilepsy ambiguously [117].
9 Potassium channels in schizophrenia and its symptoms
Numerous associations have been made between potassium channels and schizophrenia. For example, hypokalemia, consisting of low potassium, increases the risk of psychosis [118, 119]. Similar to excessive potassium channel opening, hypokalemia could lead to low potassium in membranes. A variation of the KCNH2 gene, which encodes potassium channels, has been associated with schizophrenia [120, 121]. Healthy individuals with the same genetic variant perform significantly worse cognitive wise and have a smaller hippocampus [120, 121]. This is consistent with potassium channels being involved in psychosis and cognitive impairment. Kv3 potassium channels regulate GABA interneurons. Knowing that those could contribute to schizophrenia, Kv3 modulators have been shown by early data to improve dopaminergic dysfunction in schizophrenia for both positive and negative symptom amelioration [16]. Additionally, Kv3 potassium channels have been shown to be reduced in schizophrenia and normalized after antipsychotic treatment [122]. This reduction could be explained by excessive potassium channel opening in schizophrenia leading to downregulation, while antipsychotics prevent excessive opening and regulate those channels as a result back up [29]. Indeed, antipsychotics inhibit potassium channels to a certain extent and show similar intracranially recorded ‘local field potentials’ as potassium channel modulators [123, 124]. A study using whole-cell patch-clamp recordings demonstrated the overexpression of Kv1.1, Kv1.2, Kv1.3, Kv1.6, Kv4.2, Kv4.3, and Kv7.2 channels in peripheral blood mononuclear cells of patients with schizophrenia. B lymphocytes exhibited increased output current, peak current amplitude, and voltage conductance curves among patients with schizophrenia compared with healthy controls [125]. Similarly, patch clamp recordings in a mouse model of schizophrenia demonstrated a consistent increase in the expression of small conductance calcium-activated potassium channel 3 in the PFC that contributes to enhanced medium after-hyperpolarization [126]. Those whole-cell patch-clamp findings align with the theory that schizophrenia could benefit from potassium channel closure.
9.1 Cognitive symptoms
Schizophrenia’s cognitive symptoms affect various cognitive domains [5], and potassium channel blockers could improve those. For example, inhibiting KCNQ2 and other potassium channels can improve working memory [29, 48, 62, 127]. KCNH2 variability contributes to inefficient brain activity modulation during the N-back task which measures working memory and attention in schizophrenia patients [128]. Additionally, a potassium channel gene, namely KCNQ1, has been associated with myelin and processing speed deficits in schizophrenia. The allele associated with higher expression and thus higher potassium channel activity is associated with myelin deficits, processing speed deficits, and increased risk of schizophrenia. It leads to reduced prefrontal excitability, which KCNQ1 antagonists reverse [129]. Another potassium channel gene, namely KCNH2, is associated with schizophrenia, lower I.Q., processing speed, attentional and working memory deficits [120]. Similarly, inhibiting potassium channels can improve attention and sensory gating [18, 19, 29, 127, 130]. For example, guanfacine, that closes HCN and potassium channels via increased a2-AR and decreased αΙ-AR activity, resolves sensory gating deficits [131]. Inhibiting Kv7 channels alleviates sensory gating and cognitive deficits induced by NMDA receptor blockade [61]. Lastly, inhibiting Kvl.1, Kvl.3, and other potassium channels can improve memory [29, 100, 132, 133]. Overexpression of the potassium channel gene KCNH2-3.1 is associated with cognitive impairment and schizophrenia [134]. It comes with memory and working memory deficits that are rescued by suppressing KCNH2-3.1 [134]. Additionally, antipsychotics can increase the risk of developing type 2 diabetes [10], and potassium channel blockers could prevent the associated cognitive dysfunction [96].
9.2 Disorganization and negative symptoms
Disorganization is prevalent in schizophrenia and seems related to its cognitive deficits [135]. Thus, by treating the cognitive symptoms of schizophrenia via potassium channel closure, the disorganization symptoms should improve too. Mediating disorganization, executive dysfunction may be a common symptom between ADHD and schizophrenia [5]. This executive dysfunction would lead to similar symptoms between ADHD and schizophrenia, such as disorganization [136], working memory deficits [137], slowed processing speed [138], and attentional impairment [136]. In theory, potassium channels could also have implications for attention-deficit/hyperactivity disorder (ADHD). Via low tonic dopamine/ norepinephrine, ADHD individuals could suffer from low D1/a2-AR activation and thus low potassium channel closure, leading to high potassium channel opening and thus PFC deactivation, which would lead to the executive dysfunction found in ADHD. Indeed, genetic analysis found associations between ADHD and potassium channel genes [139]. Moreover, KCNH3 potassium channel blockers have been shown to be highly effective in ADHD [130]. This means that disorganization and the other symptoms commonly found in both disorders could, in the end, be related to excessive potassium channel opening. In addition, having ADHD increases the risk of developing schizophrenia as the low tonic dopamine found in this disorder can lead to high phasic dopamine [140, 141]. Certain patients with schizophrenia who tend to be treatment-resistant show low dopamine synthesis [142], which indeed would translate to low tonic dopamine. ADHD stimulants used at therapeutic doses can improve schizophrenia’s negative symptoms without worsening psychosis by increasing tonic dopamine and thus dopamine autoreceptor activity, subsequently lowering phasic dopamine [140]. A certain schizophrenia subtype may thus consist of low tonic dopamine, leading to negative symptoms, and high phasic dopamine, leading to positive symptoms. KCNH3 potassium channel blockers are highly effective in ADHD and thus could improve, in schizophrenia, the negative symptoms and common symptoms of ADHD and schizophrenia, such as cognitive deficits and disorganization [130].
Amisulpride is the best current treatment for negative symptoms and improves those via presynaptic dopamine D2 autoreceptor blockade [143, 144]. As Kv1.2, Kv1.3, Kv1.6, and other potassium channels act downstream of presynaptic dopamine D2 as autoreceptors [52], blocking those would also disinhibit dopamine and improve negative symptoms. In addition, cognitive symptoms could contribute to negative symptoms and thus treating cognitive symptoms via potassium channel blockade may subsequently also contribute to improved negative symptoms [3]. Similarly, stimulants can improve cognition and increase dopamine. As explained above dopamine enhancing medications such as ADHD stimulants can be added safely in schizophrenia as long as the patient’s positive symptoms are under control with an antipsychotic treatment. Antipsychotics and ADHD stimulants at therapeutic doses may even act synergistically by both lowering phasic and increasing tonic dopamine [145, 146]. Blocking
KCNH3 is effective in ADHD and increases dopamine and acetylcholine [130]. As detailed above, increasing acetylcholine could reverse the low M1 activity in schizophrenia, while increasing dopamine could improve negative symptoms in schizophrenia. Thus, potassium channel blockade of KCNH3, Kv1.2, Kv1.3 or Kv1.6 could improve schizophrenia negative symptoms.
9.3 Positive symptoms
In contrast to this article’s theory, some evidence indicates potassium channel opening to augment conventional antipsychotic’s therapeutic benefit against psychosis by further lowering dopamine. However, the evidence is low and becomes ambiguous when looking at cognitive symptoms and sensory gating [18, 19]. Plus, the used potassium channel opening drugs act on additional targets that can also lower dopamine to provide an antipsychotic effect [147, 148]. On the other hand, retigabine, a potassium channel opener previously used in epilepsy, induces side effects reminiscent of schizophrenia, such as cognitive deficits, confusion, abnormal thinking, aggression, attentional problems, and even psychosis [29]. This article concludes, opening certain potassium channels could augment conventional antipsychotics by further lowering dopamine. However, opening potassium channels also seems to be associated with cognitive deficits and even other symptoms, such as aggression, found in schizophrenia. Potassium channel openers have even shown psychosis as a side effect. When looking at this article’s theory, lowering dopamine via certain open potassium channels could indeed limit ‘hyper-salience’ leading to delusions from striatum hyper-activation and ‘self-generated sensory activity’ from sensory cortex hyper-activation leading to hallucinations. On the other hand, open potassium channels would worsen the ‘cognitive impairment’/’loss of insight’/’lack of deliberate reasoning’/’lack of reality monitoring’ from an ‘offline’ PFC aspect of psychosis and, in general, worsen the cognitive and negative symptoms of schizophrenia. This means potassium channel opening is a non-optimal way of lowering psychosis risk and treating schizophrenia. Instead, closing potassium channels could improve the ‘cognitive impairment’/’loss of insight’/’lack of deliberate reasoning’/’lack of reality monitoring’ aspect of psychosis next to the cognitive and negative symptoms of schizophrenia while being complementary to conventional antipsychotics which treat the other two factors implicated in psychosis, namely ‘hyper-salience’ and ‘self-generated sensory activity’.
10 HCN channels in schizophrenia and its symptoms
Next to potassium channels, open HCN channels can also bring the PFC ‘offline’ and thus contribute to the ‘cognitive impairment’/’loss of insight’/’lack of deliberate reasoning’/’lack of reality monitoring’ factor of psychosis [29]. Consistent with this article’s theory on HCN channels contributing to schizophrenia and its cognitive symptoms, a genetic analysis found an association between the HCN1 gene and schizophrenia [149]. The genetic variant associated with schizophrenia also seems to impact working memory negatively in those schizophrenia patients [149]. In theory this could be explained by HCN channels modulating neurodegeneration and cognition. As explained above in the ‘neurodegeneration’ section, blocking HCN1 channels could not only prevent stress-induced PFC neurodegeneration but also reverse it and thus treat, or at least improve, the neurodegenerative aspect of schizophrenia. Patients with high dopamine who block D2 receptors may experience excessive HCN opening via D1 activation opening HCN channels and D2 antagonism not closing HCN channels [29], which would contribute to cognitive dysfunction and neurodegeneration. This is why D1 improves working memory in an inverted U curve [150]. The other HCN channel subtypes could also impact schizophrenia by modulating midbrain dopamine neurons [151], in particular HCN2-HCN4 subtypes [151]. Some evidence indicates that blocking HCN channels reduces the spontaneous firing activity of dopamine neurons [151]. Moreover, it has been hypothesized that HCN channels regulate phasic dopamine over tonic dopamine [151], as would be desired in the treatment of psychosis. However, lowering phasic dopamine could worsen the negative symptoms of schizophrenia.
11 Oxytocin on ion channels
Oxytocin could improve positive, negative, and cognitive symptoms in schizophrenia. But also enhance social cognition [152, 153]. Generally, oxytocin increases dopamine, but sometimes it could decrease dopamine, thus its effects on dopamine are ambiguous [154]. On the other hand, oxytocin blocks potassium and HCN channels [155, 156]. Following this article’s theory, this may explain how oxytocin positively affects all schizophrenia symptoms.
Furthermore, inhibition of Kv, KCNK2, ATP-sensitive potassium channel (KATP), KCNQ, and KCNH1-3 potassium channels increases oxytocin [157-159]. However, HCN3 channel inhibition lowers oxytocin [160]. As explained previously, HCN1 and HCN2 are of higher importance in schizophrenia, thus HCN3 could be left out as a target. On the other hand, closing potassium channels to increase oxytocin can only be an additional positive.
12 Nicotine on ion channels
Smoking is most prevalent in patients with schizophrenia compared to other patient groups and may be a way of self-medicating [161]. Nicotine improves negative symptoms and cognition in schizophrenia and patients at high risk of psychosis [161-163]. Nicotine does not increase the risk of psychosis while increasing dopamine and norepinephrine [161, 164-167], which is not consistent with the theory that excessive dopamine D2 activity is the sole contributing factor in psychosis. On the other hand, nicotine inhibits various potassium channels independently from its actions on acetylcholine receptors [168-172]. Nicotine could also block HCN and sodium channels independently from its actions on acetylcholine receptors [173-175], which is consistent with it improving ‘information maintenance’ and thus cognition. Varenicline acts on nicotinic receptors too but not on ion channels. Varenicline is not associated with similar cognitive improvements in schizophrenia and increases the risk of psychosis compared to nicotine [164-167, 176]. While nicotine benefits schizophrenia and closes potassium and HCN channels, varenicline does not. This is aligned with this article’s theory on potassium and HCN channel closure improving schizophrenia.
13 Ion channels in treatment-resistant schizophrenia
Treatment-resistant schizophrenia (TRS) refers to patients who continue to have symptoms and poor outcomes after failure of at least two antipsychotics. One-fifth to one-half of patients with schizophrenia have TRS and 30-60% of those patients respond to clozapine [1]. Clozapine is not used as a first-line treatment in schizophrenia because it comes with the risk of developing agranulocytosis [177]. We still do not know what mechanism of action behind clozapine makes it more effective than other antipsychotics [178]. However, ion channels may be involved. As explained in the ‘HCN in schizophrenia’ section above, excessive D1 activity can open HCN channels to cause cognitive dysfunction and prefrontal neurodegeneration. As positive symptoms of schizophrenia are related to high dopamine levels, high dopamine D1 activity could become problematic in certain patients. Clozapine blocks D1 and subsequently prevents excessive HCN channel opening in patients with high dopamine acting on this receptor [179]. In addition, clozapine directly inhibits potassium channels, namely the Kv11.1, Kv7.1, Kv4.3, and Kir channels [180, 181]. Potassium and HCN channel blockade, in addition to the conventional antipsychotic’s mechanisms of action, such as 5HT2A and D2 blockade, may explain clozapine’s superior efficacy. Clozapine mimics, to some extent, the treatment goal proposed in this article consisting of current antipsychotic treatments being augmented with potassium/HCN channel blockers. Lastly, clozapine could improve TRS by increasing activity on NMDA receptors [142]. As explained in the ‘glutamate’ section, NMDA receptors keep the PFC ‘online’. As a result, this is also consistent with the theory that an ‘offline’ PFC contributes to schizophrenia symptoms. To conclude, TRS patients may suffer less from hyper-dopaminergic activity in the striatum. Instead, an ‘offline’ PFC may be the major contributing factor to their psychoses, which clozapine would treat via modulation of HCN, potassium channel, and NMDA activity. Indeed, dopamine synthesis capacity in the striatum has been shown to be lower in patients with TRS who are clozapine- responsive compared to patients without TRS and even healthy controls [142]. As current antipsychotics target high striatal dopamine, it makes sense that those patients become treatment-resistant. This is consistent with the theory that high striatal dopamine is not the only factor that can lead to psychosis. The following findings could explain the association between low striatal dopamine and the PFC being ‘offline’. Catechol-O-methyltransferase (COMT) is an enzyme responsible for the removal of dopamine and norepinephrine in the PFC. Genetic alleles related to low COMT activity lead to higher PFC dopamine and norepinephrine levels, especially in women, as COMT is responsible for estradiol removal too. In women, having the genetic alleles leading to higher COMT activity, and thus lower prefrontal dopamine and norepinephrine levels, decreases the risk of TRS [182]. Higher prefrontal dopamine is associated with lower striatal dopamine [182]. Lower striatal dopamine is associated with TRS and response to clozapine. Excessive prefrontal dopamine and norepinephrine could lead to potassium/HCN channel opening, putting the PFC ‘offline’ and causing psychosis via this mechanism that conventional antipsychotics do not treat but clozapine does treat. This is consistent with this article’s theory whereby open potassium/HCN channels can contribute to psychosis too by impairing the PFC.
In addition, neuroinflammation and oxidative stress could contribute to TRS [142]. As explained in the ‘neurodegeneration’ section, conventional antipsychotics can increase oxidative stress, while blocking certain potassium channels can lower neuroinflammation and oxidative stress. HCN2 blockade can also provide an anti-inflammatory effect. Thus potassium/HCN channel blockers could also treat TRS related to neuroinflammation and oxidative stress.
Lastly, TRS due to dissociation could also benefit from potassium/HCN channel blockers. Dissociation is associated with a loss of integrity between memories and perceptions of reality. A strong relationship exists between the positive symptoms of schizophrenia and dissociation. Patients with TRS experience twice as much dissociation compared to patients in remission. Moreover, those TRS patients experience sufficient dissociative symptoms for a dissociative personality disorder diagnosis. As previously theorized, certain forms of psychosis may be dissociative at the core and would benefit from interventions targeting dissociation [183, 184]. Recent evidence indicates that HCN1 channel opening is required for NMDA antagonists to induce dissociative states [185]. Interestingly, the kappa-opioid agonism pathway is the only other psychosis-related pathway that causes dissociation [2, 178], and it is known to lead to high Kir potassium channel opening, as explained in the ‘kappa-opioid’ section above. Thus, closing HCN1 and Kir channels may provide an anti-dissociative effect that may improve psychosis in a subset of patients who are resistant to conventional antipsychotics.
14 Discussion
Following the literature review, the following theory has not been disproven; the reviewed evidence even suggests it to be true. An ‘offline’ PFC from potassium and HCN channel opening leading to ‘cognitive impairment’/’loss of insight’/’lack of deliberate reasoning’/’lack of reality monitoring’ may be one contributing factor in psychosis. Next to ‘hyper-salience’ leading to delusions from striatum hyper-activation and ‘self-generated sensory activity’ leading to hallucinations from hyper-activation of the sensory cortex. These contributing factors may converge to varying degrees to, ultimately, result in psychosis. This article’s theory could explain how kappa-opioid agonists and potassium channel openers induce psychosis while lowering dopamine but opening potassium channels; how nicotine improves certain schizophrenia symptoms while increasing dopamine but closing potassium/ HCN channels; how insightfulness is maintained with 5HT2A psychedelics which increase dopamine but close potassium channels; why guanfacine which closes potassium/HCN channels is the best treatment in delirium psychosis which is characterized by prominent cognitive dysfunction; and why clozapine which closes potassium/HCN channels is superior to other antipsychotics.
However, after the literature review, on top of the contemplated theory, it can be added that NMDA receptor activation also plays a role in keeping the PFC ‘online’. In addition to addressing PFC impairment, increasing NMDA activity improves the hyperactivation of the striatum and sensory cortex in psychosis by lowering dopamine. Thus, increasing NMDA activity could theoretically provide positive and cognitive symptom improvement. HCN channel closure could maybe lower phasic dopamine and thus improve the striatum and sensory cortex hyper-activation aspects of psychosis too. In addition, HCN channel closure, and potassium channel closure to a lesser degree, could rescue the neurodegenerative aspect behind schizophrenia. On the other hand, potassium channel closure, and HCN channel closure to a lesser degree, could improve schizophrenia’s negative symptoms. Finally, after the literature review, it can be added that not only high dopamine in the striatum can contribute to the ‘hyper-salience’ leading to delusions aspect of schizophrenia but probably high norepinephrine in the PFC too and that high norepinephrine would especially lead to paranoid delusions.
Fundamentally, this article’s theory is based on prefrontal dysfunction, and at its core, prefrontal dysfunction arises from reduced neuronal excitability in this region. Increasing NMDA activity and closing potassium and HCN channels are different methods for enhancing excitability. Even if NMDA receptor activation already plays a role in keeping the PFC ‘online’, patients may still benefit from potassium or HCN channel blockers instead. Blocking those channels may offer additional benefits by reversing neurodegeneration and alleviating negative
symptoms.
To conclude, as a simplification, this article theorizes that having a cognitive deficit in the first place may make someone more susceptible to developing all schizophrenia symptoms and that potassium/HCN channel blockers would improve that. In contrast, as a comprehensive extension, Figure 1 illustrates all the pathways contributing to schizophrenia and psychosis as theorized in this article.
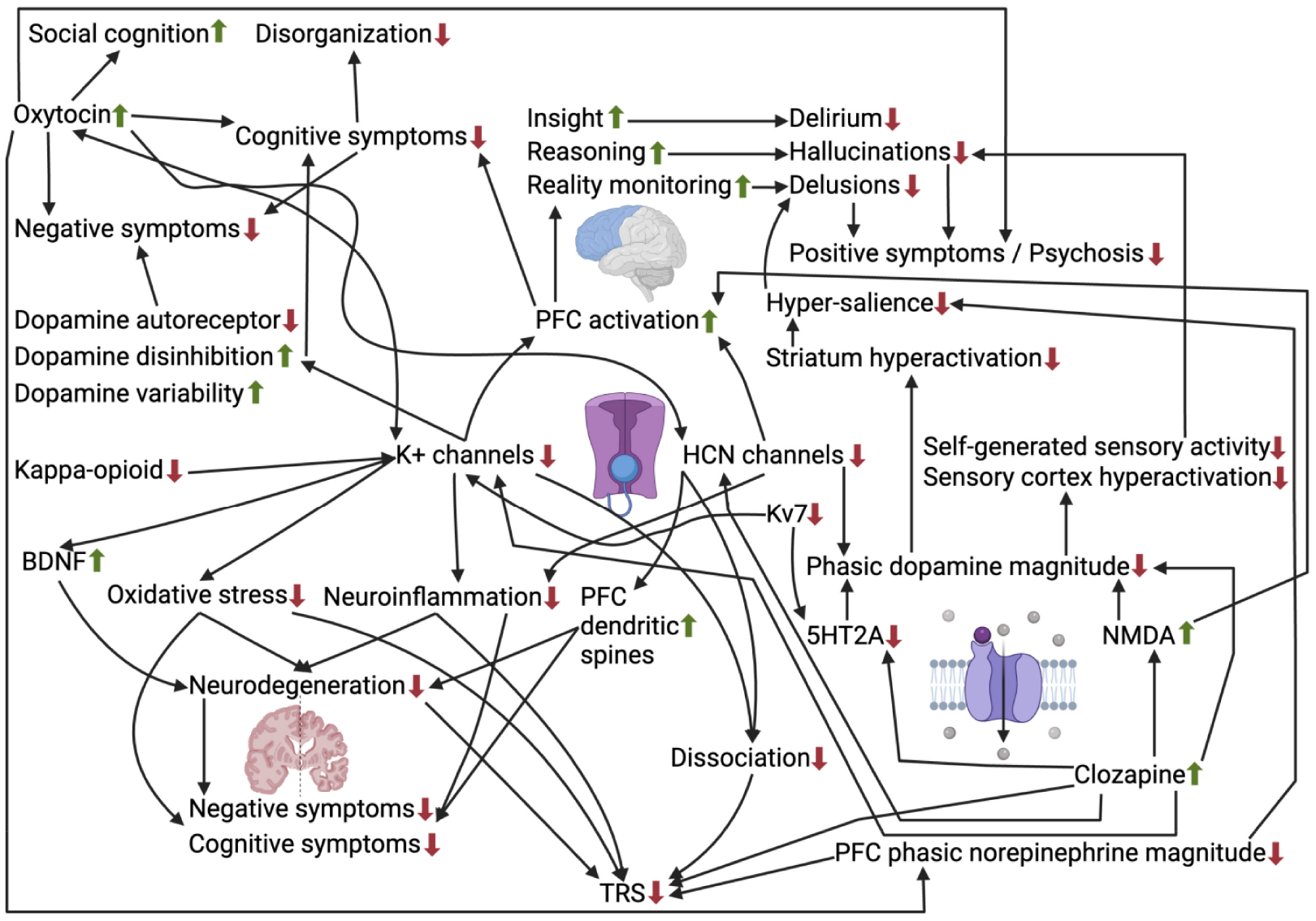
Visual representation of pathways, including potassium and HCN channel closure, impacting psychosis and the various schizophrenia symptoms.
14.1 Treatment perspectives
Before presenting treatments, it is important to know who would or would not benefit from the proposed treatments. To evaluate this, first, it is of interest to consider what channel benefits which disorders/symptoms. This article theorizes that agents closing potassium and HCN channels would particularly benefit cognitive and negative symptoms of schizophrenia; positive symptoms to a lesser extent to reduce but not replace the need for conventional antipsychotics. Additionally, this article theorizes that delirium psychosis is mainly due to cognitive dysfunction and thus that potassium/HCN channel blockers could completely replace conventional antipsychotics in its treatment. To be more precise, a cognitive test could be used to predict what patient groups would benefit from potassium/HCN channel blockers. The following presents a speculative hypothesis regarding a cognitive test to measure PFC dysfunction in patients with psychosis. When open, potassium channels allow positive ions to exit the membrane, while HCN channels permit positive ions to enter. Both channels, when opened, lead to membrane charge updating which may correspond to ‘information updating’ at the detriment of ‘information maintenance’. Excessive opening of those channels could lead to a lack of ‘ information maintenance’ and thus cognitive impairment. Consistent with the idea of patients with schizophrenia lacking ‘information maintenance’, they appear more sensitive to recent information [25], as if they forgot prior information. Working memory may directly measure ‘information maintenance’ [50], and indeed, a deficit in working memory is found in schizophrenia [5]. This article hypothesizes that in a healthy brain, information is maintained in working memory via tonic/moderate excitatory neurotransmission at baseline and becomes updated when salient stimuli comes along and activates phasic/high excitatory neurotransmission to downstream open potassium/HCN channels. In psychotic disorders, an excess of stimuli would be considered salient, leading to excessive ‘information updating’ and insufficient ‘information maintenance’ due to overactivation of potassium/ HCN channels. As a result, ‘information maintenance’ could serve as a cognitive measure estimating PFC dysfunction in patients with psychosis, and working memory may be a simple measurement reflecting that. Low working memory function would be an indication for potassium/HCN channel blockers in patients with psychosis. Cognitive testing could pave the way to personalized treatments for psychiatric disorders.
As a treatment, potassium/HCN channels could be the direct target, or indirect target by acting on upstream neurotransmitters first. As previously mentioned, both high or low levels of various neurotransmitters can, ultimately, lead to potassium/HCN channel modulation. High dopamine levels in the striatum or sensory cortex appears almost universal in schizophrenia and leads to ‘hyper-salience’ or ‘self-generated sensory activity’, respectively. Nevertheless, other neurotransmitters could be out of balance for the prefrontal cortex to become ‘offline’ and cause ‘cognitive impairment’/’loss of insight’/’lack of deliberate reasoning’/’lack of reality monitoring’. Directly modulating potassium/HCN channels would be easier than correcting the different upstream neurotransmitter systems of each patient in a personalized manner. As a result, potassium/HCN channels as the direct target would be the desired treatment. However, direct potassium and HCN channel blockade is not devoid of undesirable potential side effects. Blocking repolarizing Kv11.1 and Kv7.1 potassium channels can increase the risk of potentially fatal Torsades de Pointes. Simultaneously blocking depolarizing CaV1.2 calcium channels and NaV1.5 sodium channels leads to mutual compensation. Clozapine blocks these channels simultaneously and has a satisfactory arrhythmogenic safety profile [180]. Potassium channel blockers may also increase prolactin, and calcium channel blockers could prevent this effect [186]. Therefore, simultaneous calcium channel blockade could prevent side effects from potassium channel blockade. Similarly, the blockade of HCN channels also increases the risk of Torsades de Pointes [187]. Conversely, guanfacine lowers prolactin and improves cardiovascular health while inhibiting potassium and HCN channels [186, 188]. As discussed earlier in the ‘5HT2A’ section, guanfacine has been shown to be highly effective in delirium psychosis, which is primarily characterized by prefrontal dysfunction and shows poor response to conventional antipsychotics [71]. Guanfacine is also used off-label to treat cognitive deficits in schizotypal patients [45, 124]. A meta-analysis demonstrated guanfacine to have some efficacy as a cognitive enhancer in schizophrenia [189]. In contrast, atomoxetine and reboxetine, which increase norepinephrine with a lower a2-AR to a1-AR ratio and therefore close potassium and HCN channels less, have not been shown efficacious [190]. A previous case report demonstrated improvements in schizophrenia’s residual symptoms with guanfacine in a patient with comorbid ADHD [191]. As discussed earlier, guanfacine blocks HCN channels by modulating norepinephrine receptors, subsequently lowering cAMP, which mainly lowers HCN2 and HCN4 while HCN1 only to a lesser extent. This makes guanfacine suboptimal on its own. Lastly, selective blockade of HCN1 can result in phosphenes as a side effect [102]. Clozapine can reduce HCN activity without associated side effects through D1 receptor blockade, as explained in the ‘treatment-resistant schizophrenia’ section. Other indirect methods exist to close potassium and HCN channels while avoiding undesirable side effects. Inhibiting cAMP-PKA signaling is one method to close potassium and HCN channels [29]. Opioids which can provide an antipsychotic effect while increasing dopamine, as discussed in the ‘norepinephrine’ section, indeed inhibit cAMP-PKA. Additionally, genetic associations with cAMP in schizophrenia have been made [127]. However, cAMP is essential for long-term learning-related synaptic plasticity [192]. Furthermore, cAMP inhibition mainly lowers HCN2 and HCN4, while it only slightly lowers HCN1 [102]. As a result, cAMP inhibition may not suffice on its own as previously noted when contemplating guanfacine. Alternatively, blocking phosphatidylinositol 4,5-bisphosphate (PIP2) can reduce HCN channels [151], and may also lower the opening of KCNQ among other potassium channels [193]. Similarly, blocking p38 mitogen-activated protein kinases (MAPK) can reduce HCN channels [151]. Inhibition of the MAPK pathway could also lower Kv potassium channels [194]. MAPK inhibitors are commonly used in the treatment of inflammatory diseases and cancer [195]. Nevertheless, they have been hypothesized to be of use in psychiatric stress disorders such as anxiety, depression, and addiction [196]. Additionally, they could prevent adverse effects from kappa-opioid activation and increase AMPA receptor expression [196], which downstream increases NMDA activity [197]. Increased NMDA activity would also improve PFC impairment. To conclude, it remains to be tested if cAMP, PIP2, or MAPK inhibitors, alone or together, are sufficient in their ability to close HCN and potassium channels to provide a therapeutic effect.
It is also worth mentioning how available potassium channel blockers have been shown to affect schizophrenia symptoms. The specific Kv7 channel blocker XE991 has been reported to alleviate cognitive deficits in an animal model of schizophrenia cognitive symptoms [61]. Furthermore, it acts downstream of dopamine D2 autoreceptors to disinhibit dopamine [198], potentially improving negative symptoms in a manner similar to amisulpride, as discussed in the ‘disorganization and negative symptoms’ section. However, by increasing dopamine, XE991 has been shown to worsen an animal model of schizophrenia positive symptoms [199]. Tipepidine, an over-the-counter nonnarcotic antitussive used in Japan since 1959 [200], inhibits G-protein-coupled inwardly rectifying potassium (GIRK)-channel currents [200]. It increases dopamine by inhibiting D2 receptor-mediated potassium channel currents [201]. As discussed earlier in the ‘disorganization and negative symptoms’ section, this suggests that tipepidine can improve the negative symptoms of schizophrenia similarly to amisulpride. Furthermore, in a mouse model of schizophrenialike cognitive dysfunction, tipepidine elicited recovery from the induced cognitive impairment [202]. It also attenuated an animal model of schizophrenia positive symptoms [202]. The safety of tipepidine has already been established in both children and adults [200]. Therefore, the safe and available tipepidine holds potential to improve all schizophrenia symptoms.
A distinction can be made between different subtypes or upstream mediators of potassium and HCN channels. Table 1 summarizes these distinctions to guide precise, targeted treatment.
Summarizes what is currently known about the different ion channel subtypes and their upstream mediators in relation to psychosis and schizophrenia, as reviewed and theorized in this article

14.2 Limitations
The limitations of this study are that data is sparse on how selective potassium and HCN channel modulating drugs affect psychosis and schizophrenia directly. This results in a theory primarily based on inferential reasoning. Certain axioms used for inferences related to schizophrenia in humans come from animal or “in vitro” studies. The regional distributions of neurotransmitters and channels influencing schizophrenia symptoms are not always specified. Table 1 is likely incomplete, as little is known about the various subtypes of potassium/HCN channels.
14. 3 Directions for future research
The following studies could verify if potassium and HCN channel openers induce psychosis and/or other symptoms of schizophrenia. This could validate the theory that open potassium and HCN channels lead to PFC dysfunction followed by ‘cognitive impairment’/’loss of insight’/ ‘lack of deliberate reasoning’/’lack of reality monitoring’ to, ultimately, cause psychosis and/or other schizophrenia symptoms. Subsequently, delirium psychosis can be studied in relation to potassium/HCN channel blockers. Delirium psychosis is a form of psychosis with high PFC dysfunction that tends to worsen as cognition symptoms escalate [70, 71]. As a result, following our theory, it should respond the most to potassium and HCN channel blockers. Next studies could compare potassium and HCN channel blockers to conventional antipsychotics in the treatment of delirium psychosis. This could validate the theory that delirium psychosis is mainly due to cognitive dysfunction and that potassium/HCN channel blockers are superior to conventional antipsychotics in its treatment. If previously suggested studies validate the theory surrounding potassium and HCN channels being able to play a role in cognitive dysfunction and psychosis, then, next studies could directly evaluate it in schizophrenia patients. More specifically, cognitive, negative and positive schizophrenia symptoms could be evaluated after the addition of potassium/HCN channel blockers to a conventional antipsychotic other than clozapine. This could validate the theory that agents closing potassium and HCN channels would especially benefit cognitive and negative symptoms of schizophrenia while being complementary in schizophrenia as an add-on treatment to conventional antipsychotics who mainly treat positive symptoms. Lastly, if potassium/HCN channel blockers turn out to help at least certain schizophrenia patients, a future study could evaluate the treatment response relative to a cognitive test measuring working memory. This could validate the theory that poor working memory is related to poor information maintenance which is related to open potassium/HCN channels. Working memory could then be used in practice as an indication for treatment with potassium/HCN channel blockers or not. Nevertheless, side effects such as Torsades de Pointes [180], seizures [114], and hyperprolactinemia should be monitored when using potassium channel blockers [186]. Similarly, when using HCN channel blockers, Torsades de Pointes should also be monitored next to phosphenes [102, 187].
Footnotes
Acknowledgements
None.