
Abstract
The Hippo pathway, a highly conserved kinase cascade, regulates cell proliferation, apoptosis, organ size, and tissue homeostasis. Dysregulation of this pathway reportedly plays an important role in the progression of various human cancers. Yes-association protein (YAP), the Hippo pathway’s core effector, is considered a marker for cancer therapy and patient prognosis. In addition, studies have indicated that YAP is involved in promoting anticancer drug resistance. This review summarizes current knowledge on YAP’s role in cancer progression, anticancer drug resistance, and advances in the development of YAP-targeting drugs. A thorough understanding of the complex interactions among molecular, cellular, and environmental factors concerning YAP function in cancer progression may provide new insight into the underlying mechanism of anticancer drug resistance. It might lead to improved prognosis through novel combined therapies.
1 Introduction
The Hippo pathway is a highly conserved signaling pathway originally discovered in Drosophila [1–3]. In mammals, the core components of the classical Hippo pathway include mammalian sterile 20-like protein kinase 1/2 (MST1/2), large tumor suppressor 1/2 (LATS1/2), Yes-association protein (YAP) or transcriptional co-activator with PDZ binding motif (TAZ), and TEA domain-containing transcription factor (TEAD). Upon activation of the Hippo pathway, MST1/2 is phosphorylated. Then, it phosphorylates two adaptor proteins, Salvador homolog1 (SAV1) and Mob kinase activator 1 (Mob1). SAV1 recruits MST to the membrane to promote its interaction with other proteins. Mob1 regulates the interaction between MST1/2 and its downstream kinase, LATS1/2. LATS1 is phosphorylated at Thr1079, and LATS2 is phosphorylated at Thr1041 by MST1/2, together with the adaptor proteins. Furthermore, LATS phosphorylated YAP at different sites, resulting in a different fate. When LATS1/2 phosphorylates YAP at Ser127 in mammals (Yki at Ser168 in Drosophila), a binding site for 14-3-3 is created, which results in the cytoplasmic retention of YAP. When LATS1/2 phosphorylates YAP at Ser381, YAP is recognized and ubiquitylated by SCF-β-TrCP, degrading YAP [4, 5]. When the Hippo pathway is inactivated, YAP translocates into the nucleus, where it interacts with TEAD to activate the transcription of its target genes [6, 7] including connective tissue growth factor (CTGF), cysteine-rich protein 61 (CYR61) [8, 9], and anexelekto (AXL) [10–12], thus regulating cell cycle and proliferation (Fig. 1).
As a transcriptional co-activator, YAP is involved in many cellular and physiological processes, such as stemness, mechanotransduction, cell polarity, and immune suppression, many of which are considered hallmarks of cancer [13]. For example, YAP regulates the expression of transcription factors including Sox2, Nanog, and Oct4 to maintain pluripotency or stemness in human embryonic stem cells and in induced pluripotent stem cells (iPSCs) [14]. YAP is recognized as a mechanosensor within the tissue microenvironment that responds to extrinsic and cell-intrinsic mechanical signals and, therefore, regulates cell morphology and cytoskeletal tension through a mechano-activated transcriptional program [15, 16]. In a 3D spheroid model, YAP regulates tissue tension and fibronectin assembly by increasing ARHGAP18 transcription and subsequently inhibiting Rho activity [17]. Inflammation is an adaptive response triggered by stressful conditions, such as infection and tissue injury. The long-term expression of YAP in hepatocytes positively correlated to the degree of liver inflammation. Hepatocyte-specific YAP potently activates the expression of inflammatory factors, including IL-1β and TNFα [18]. Cell polarity is a fundamental entity of most cells. Studies have shown that YAP is indispensable for establishing the correct localization of polarity proteins and maintaining cell polarity. In lens cells, YAP knockout results in the loss of epithelial polarity and a change in cell shape [19].
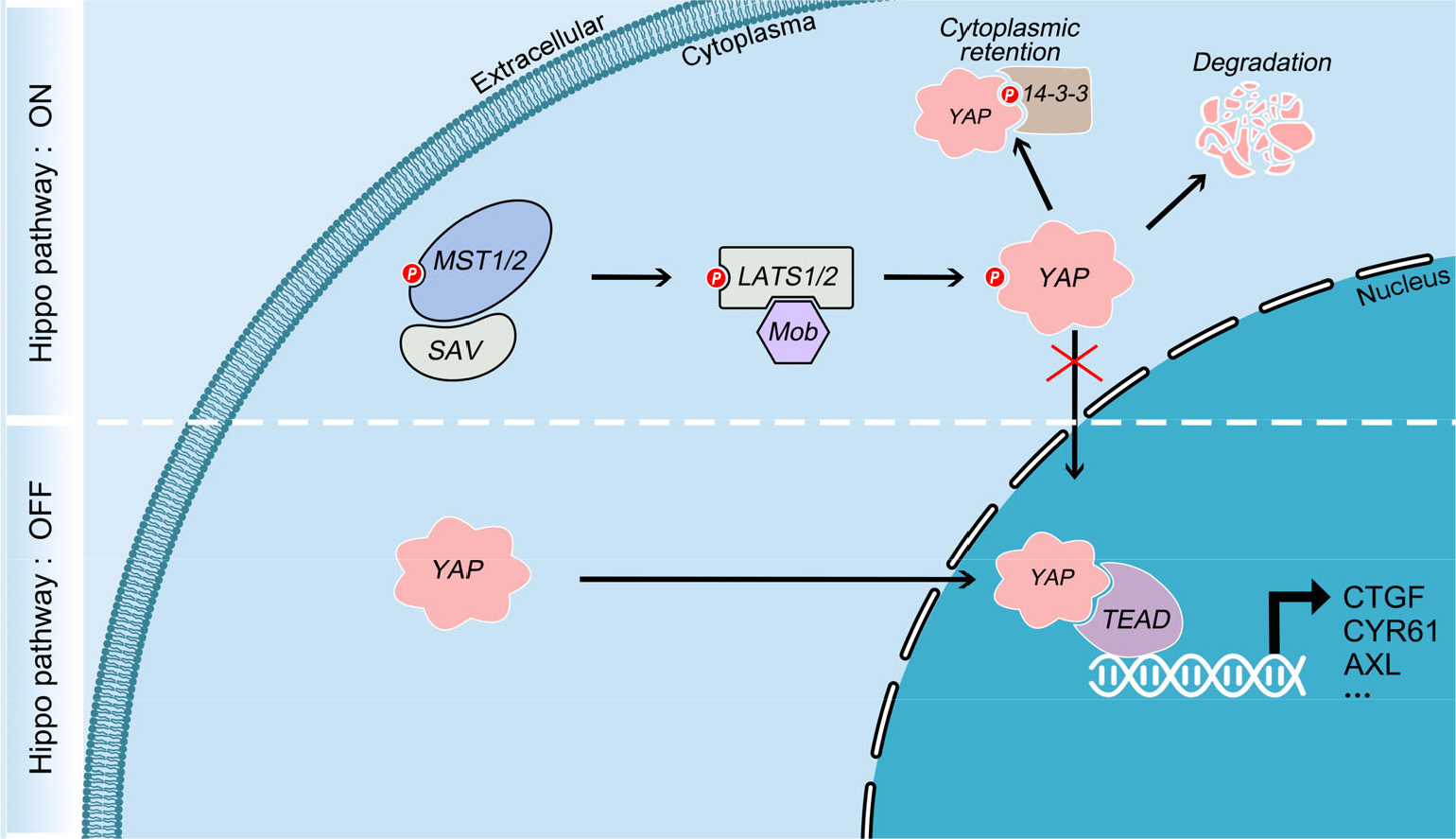
Schematic representation of core components of the Hippo pathway and their regulators in mammals. After Hippo pathway activation, the activated MST1/2-SAV complex phosphorylates and activates the LATS1/2-Mob complex, then phosphorylates YAP, resulting in its cytoplasmic retention and degradation. When the Hippo pathway is inactivated, YAP translocates into the nucleus and interacts with TEAD to activate the transcription of its target genes.
Over a decade of research revealed that YAP activity is regulated by low cell density, stiff extracellular substrates, high mechanical stress, and increased nutrient uptake [20–22]. In addition, YAP integrates various oncogenic signaling pathways inputs, including Wnt, TGFβ, KRAS, MAPK, GPCR, and shh signaling [23, 24]. YAP is also an important component of the β-catenin destructive complex and inhibits Wnt signaling by retaining β-catenin in the cytoplasm [25, 26]. YAP is suppressed by DVL, the scaffolding protein of the Wnt pathway [27]. Our previous study demonstrated that YAP regulates the activity of β-catenin by modulating GSK3β activity [28]. YAP also promotes SHP2 transport to the nucleus and ultimately activates Wnt signaling [29]. YAP binds to the Smad2/3-Smad4 complex to maintain its nuclear accumulation, and TGF-β mediates stem cell self-renewal and neural epithelial differentiation [30, 31]. With OCT4, YAP and Smad2/3 form a complex that promotes the transcription of various genes that suppress mesendodermal differentiation [32]. A recent study found that KRAS induces OTUB2 poly-SUMOylation and thereby activates YAP [33]. In Hippo dysregulated tumors, an interaction between the MAPK pathway and TEAD stability occurs [34]. The p38 kinase promotes the cytoplasmic translocation of TEAD and inhibits the growth of YAP-driven cancer cells in response to various stress signals [35]. Mounting evidence suggests that deregulation of YAP plays a significant role in cancer initiation, progression, and the promotion of anticancer drug resistance. This review focuses on the effect of YAP on cancer progression, anticancer therapy resistance, and the development of YAP-targeting drugs.
2 Function of YAP in cancer initiation and progression
Dysregulation of the Hippo pathway reportedly plays an important role in the progression of multiple cancer types. Numerous reviews describe the function of YAP in cancer initiation and progression [25, 36–38]. YAP is upregulated or hyperactivated in many solid tumors, including lung, breast, and pancreatic cancers (Table 1). In addition, higher expression of YAP is associated with poor prognosis and shorter patient survival. YAP transcriptionally activates target genes involved in cell stemness, proliferation, migration, and invasiveness as an oncogene, thus promoting cancer progression through different mechanisms.
Functions of YAP in cancer progression.

2.1 Lung cancer
Overexpression of YAP correlates with the occurrence, progression, and poor outcome associated with non-small-cell lung cancer (NSCLC) [40]. In addition, germline YAP R331W mutation may increase NSCLC risk [58]. Thus far, this YAP mutation has not been reported in other types of cancer. YAP is required to maintain the stemness properties of lung cancer cells [39], and YAP silencing results in a profound reduction of spheroid formation and the downregulation of stem cell markers [39]. A recent study reported that YAP is active in lung tumor-propagating cells with metastatic potential [59], and YAP knockdown reduced the size of tumors in mice [59, 60]. YAP’s ectopic expression in KRAS(G12D) lung cancers accelerated lung adenocarcinoma progression. Anti-apoptotic proteins as mediators of YAP are responsible for promoting the malignant progression of adenocarcinomas [60]. In adenocarcinoma and squamous cell carcinoma, high levels of YAP in the nucleus positively correlated with that of cyclin A and MAPK. They indicated tumor node metastasis (TNM) in low-stage lung squamous cell carcinoma [40].
2.2 Breast cancer
In breast cancer, elevated expression of YAP correlates with malignant tumor progression and cell proliferation, matrix stiffening, angiogenesis, migration, and invasion [61, 62]. Moreover, high expression of YAP in breast cancer cells was associated with reduced survival outcomes [63]. The interaction of YAP with KLF5 prevents E3 ubiquitin ligase from ubiquitinating KLF5, thus promoting breast cell proliferation and survival. YAP overexpression upregulates KLF5 protein levels and the expression of its target genes, including fibroblast growth factor-binding protein 1 (FGFBP1) and integrin beta-2 (ITGB2), whereas depletion of YAP decreased breast cell proliferation and survival [41]. The interaction between YAP and SMAD2/3 upregulates neuronal growth regulator 1 (NEGR1) to increase the metastatic potential of breast cancer. The interaction of nuclear YAP with TGF-β promotes phenotypic and transcriptional changes in normal breast cells, which results in the formation of cancer cells [42]. The YAP-TEAD complex induces the expression of CYR61 and CTGF, which contributes to breast cancer progression [8, 42, 43]. YAP-TEAD also binds to the promoter of the receptor for hyaluronan-mediated (RHAMM) and controls its transcription, resulting in the activation of ERK and promoting cancer metastasis [44]. Tumor suppressive effects of YAP have also been reported. Knockdown of YAP in breast cell lines suppressed anoikis, increased migration and invasiveness, inhibited the response to Taxol, and enhanced tumor growth in nude mice [64]. YAP binds to anti-apoptotic proteins to protect cells from DNA damage [65] and induces p73-mediated apoptosis following DNA damage [66].
2.3 Gastric cancer
Significant YAP upregulation occurred in the nucleus and cytoplasm of high-grade dysplasia, gastric adenocarcinoma, and metastatic gastric cancer [67]. In addition, high YAP expression reduced survival in gastric cancer patients [45, 68]. Inhibition of YAP inhibited the expression of β-catenin, c-myc, and cyclin D1 protein and increased caspase-3 and caspase-9 protein expression. Overexpression of YAP increased the proliferation of MKN45 cells [69]. Another study revealed that high expression of YAP increases c-Fos expression induced by serum and EGF treatment and promotes cell growth and motility [45]. The interaction between YAP and runt-related transcription factor 2 (RUNX2) increases oncogenic transformation by reducing p21 protein expression. Co-expression of RUNX2 and YAP significantly increased foci formation and cell growth [46]. YAP and 14-3-3ζ expression exhibited a negative correlation in gastric cancer and 14-3-3ζ promoted the phosphorylation of YAP, which resulted in its cytoplasmic retention and inactivation. YAP and 14-3-3ζ form a unique negative feedback loop in gastric cancer [68]. YAP was also reported to be negatively regulated by tumor suppressor microRNAs, such as miR-15a, miR-16-1 [47], and miR-506 [48] in gastric cancer.
2.4 Hepatocellular carcinoma (HCC)
YAP is expressed in most HCC cells and accumulates in the nucleus. Overexpression of YAP is associated with tumor differentiation and is a poor prognostic marker for HCC patients [70, 71]. CTGF is a YAP target gene that contributes to HCC cell dedifferentiation, inflammation, and cell growth by downregulating the expression of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and inhibiting TRAIL-mediated apoptosis [49]. Increased ectopic expression of YAP in HCC confers tumorigenicity by AXL, another YAP-targeting gene [10]. Upregulation of YAP activity may increase the occurrence of liver tumors [72]. Moreover, YAP reduces cellular senescence, whereas YAP downregulates, resulting in the opposite effect [51]. Cellular senescence induced by YAP deficiency depends on Rb/p16/p53. CDK6 is a direct target gene of YAP in regulating cellular senescence, and ectopic expression of CDK6 restored YAP knockdown-induced senescence [51]. YAP also interacts with various signaling pathways to promote tumorigenesis in the liver. For example, the interaction of MEK1 and YAP is important for tumor growth and maintaining the transformed phenotype of liver cancer cells [73]. Double knockout of MST1/2 in the liver induces tissue growth and HCC progression [74, 75].
2.5 Colorectal cancer (CRC)
YAP levels are associated with TNM stage and tumor status of CRC, and nuclear expression of YAP is closely associated with poor outcome [76–78]. Studies have shown that overexpression of YAP is highly associated with lymph node metastasis [79]. The siRNA-mediated knockdown of YAP expression dramatically reduced its capacity for proliferation, metastasis, and invasion [79]. Angiomotin (AMOT) upregulates activated YAP by inhibiting its phosphorylation in the cytoplasm and upregulating its expression in the nucleus. Further analysis revealed that transfection of YAP siRNA markedly reduced cell proliferation, migration, and invasion in AMOT-overexpressing CRC cells. AMOT upregulation activates ERK and AKT pathways by YAP expression, both of which are associated with the development of CRC [80]. YAP cooperates with β-catenin to promote stem cell expansion for epithelial repair, accelerating CRC growth [81]. YAP inhibits PTEN translation by activating the expression of miR-29 and the mTOR pathway to promote colon cancer growth [52]. In KRAS-overexpressing cells, upregulation of YAP restores cell viability when KRAS is inhibited. Another study demonstrated that YAP promotes the epithelial-mesenchymal transition (EMT) by activating the FOS transcription factor [53].
2.6 Pancreatic ductal adenocarcinoma (PDAC)
YAP is almost absent in the pancreas in normal tissues, but YAP nuclear expression was observed in metastases derived from PDAC [82–84]. YAP deletion inhibited the progression of early neoplastic lesions to PDAC without affecting normal pancreatic development or endocrine function. YAP functions as a central transcriptional effector to promote the expression of growth factors, which maintain tumor proliferation, which is a stromal tumorigenic response of the tumor microenvironment [85]. The interaction between the YAP/TEAD complex and the E2F transcription factor drives KRAS(G12D)-independent tumor initiation and activates the cell cycle in PDAC [54]. A recent study showed that YAP activation is enhanced in the squamous subtype and associated with a shorter overall survival time. Activation of YAP resulting from elevated WNT5A expression in progenitor subtype cancer cells significantly enhanced the malignant phenotype. YAP silencing suppressed PDAC tumorigenicity and recruited myeloid-derived suppressor cells to promote an immunosuppressive microenvironment in PDAC [86]. YAP is also negatively regulated by miR-141 [87] and miR-375 [88], which serve as independent prognostic factors in PDAC patients and function as neoplastic suppressors. Moreover, YAP expression levels strongly correlated with an myeloid-derived suppressor cells (MDSC) gene signature, and high expression of YAP predicts reduced survival in PDAC patients [86].
2.7 Glioma
We performed systematic studies on the effects of Hippo/YAP on gliomas. We initially found that the mRNA levels of YAP and its target genes, CYR61, CTGF, and BIRC5, were significantly increased in gliomas [89], which is consistent with a previous study [90]. Based on TCGA database, YAP and BIRC5 are positively correlated with the prognosis of glioma patients, whereas LATS1/2 showed a negative association with glioma patient prognosis [89]. YAP downregulation reduced glioma cell migration and invasion, whereas its upregulation caused the opposite effect [55, 89]. In addition, YAP promotes human glioma growth by activating the Wnt/β-catenin pathway [28] and N-cadherin and Twist [55]. We also demonstrated that YAP promotes glioma cell autophagy under basal and induced conditions, mediated by the high mobility group box 1 (HMGB1) [56]. Other studies in glioma cells revealed that YAP exhibits a radioresistant function by increasing DNA damage repair. Inhibition of YAP-FGF2-MAPK signaling sensitizes gliomas to radiotherapy and extends the survival of xenograft models [57]. Furthermore, the inhibition of MST1 promotes the proliferation and growth of glioma cells and inhibits apoptosis. In contrast, overexpression of MST1 reduces glioma cell growth by activating the AKT/mTOR signaling pathway but not the Hippo/YAP pathway [91], suggesting that MST1/2 may not be an upstream regulator of YAP in gliomas. LATS2 phosphorylates YAP and reduces cell growth and migration [92]. Other studies found that LATS1 mRNA and protein expression were significantly down-regulated in glioma tissues [93]. The function of ACTL6A in glioma cell proliferation, invasion, and migration is mediated by the YAP and β-TrCP E3 ubiquitin ligase interaction [94].
3 YAP in resistance to anticancer therapies
Currently, radiotherapy combined with chemotherapy represents standard methods to prolong the survival of cancer patients, with molecular targeted therapy and immunotherapy contributing in recent years. However, despite the improvement in cancer treatment, the effectiveness of anticancer therapies is limited because of intrinsic or acquired resistance. Because of the benefits of overcoming resistance to treatment, identifying anticancer therapy resistance’s underlying mechanisms has remained an important area of cancer research. In recent years, studies revealed that high YAP activity is associated with antitumor resistance to chemotherapy, molecular target therapy, radiotherapy, and immunotherapy. The mechanisms of YAP-mediated anticancer therapy resistance are presented in Fig. 2.
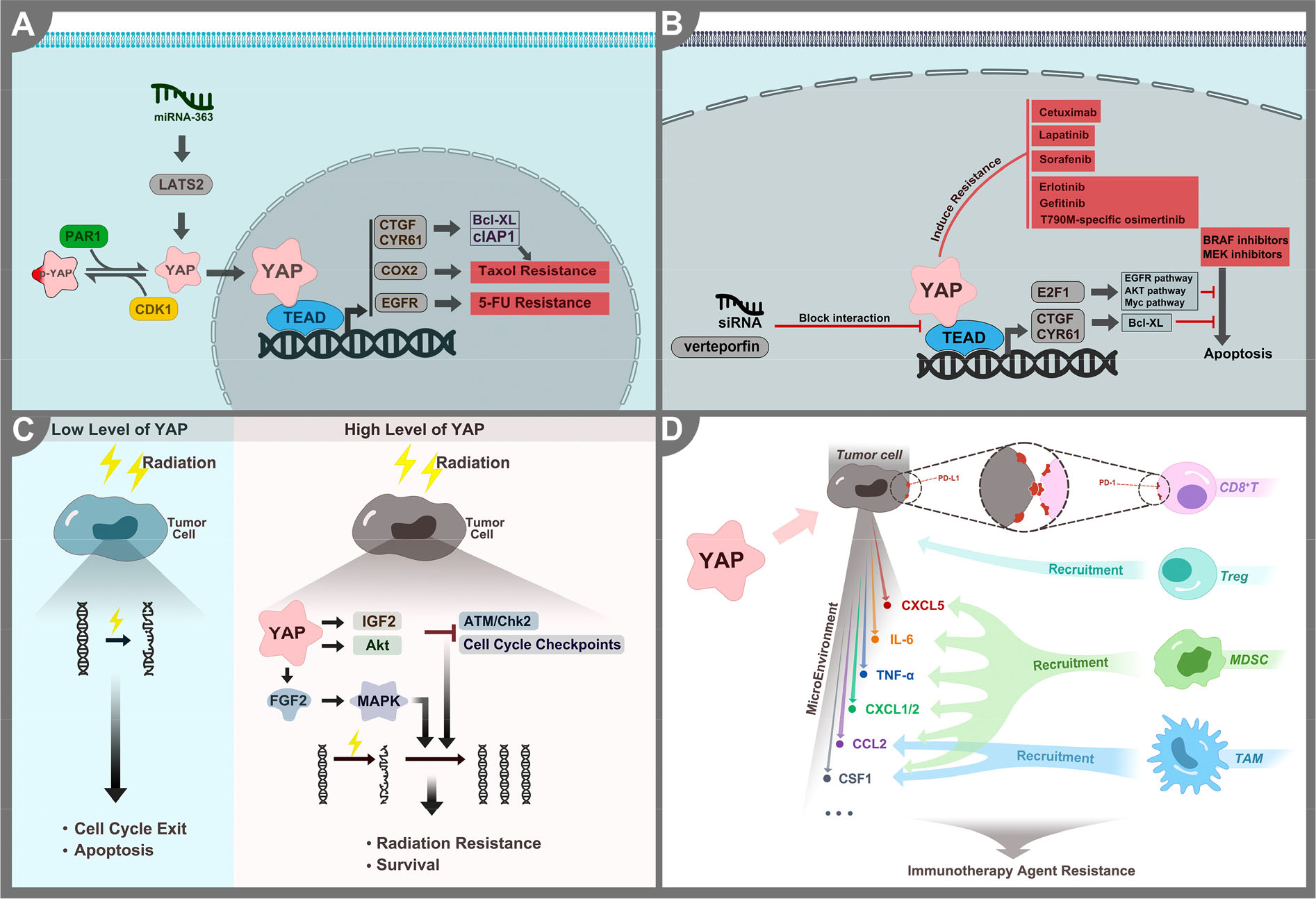
The mechanisms of YAP-mediated anticancer therapy resistance. (A) YAP promotes resistance to different types of chemotherapy by upregulating pro-survival and anti-apoptotic genes. Phosphorylating YAP sensitizes cancer cells to chemotherapy. (B) YAP promotes resistance to different targeted therapies by promoting the expression of pro-proliferative genes and anti-apoptotic genes. VP and siRNA enhance sensitivity by blocking the interaction between YAP and TEAD. (C) High levels of YAP promote resistance to radiotherapy by activating different signaling pathways to promote cell survival. In contrast, low levels of YAP induce cell cycle exit and apoptosis. (D) YAP activation promotes the expression of PD-L1 and recruits immune suppressor cells. In contrast, YAP knockout promotes the reactivation of the immune response.
3.1 Chemotherapy
Chemotherapy is the first-line therapy for most cancers. Multiple types of chemotherapy are routinely used in clinical practice, such as anti-tubulin, anti-metabolite, and DNA-damaging agents. All of these drug classes show reduced effectiveness in high YAP-expressing cancers.
The potent anti-tubulin drug, Taxol, is widely used to treat various tumor types. YAP indirectly induces Taxol resistance by inducing the expression of CTGF and CYR61, which leads to the inhibition of Taxol-induced apoptosis by upregulating the anti-apoptotic proteins, Bcl-XL, and clAP1, and promoting survival [95–98]. CDK1 reduces YAP’s stability and transcriptional activity through phosphorylation, thus sensitizing ovarian cancer cells to Taxol [99]. Activation of PAR1 induces stemness and Taxol resistance by promoting YAP dephosphorylation and its nuclear translocation in gastric cancer cells [100]. COX-2 expression induced by YAP increases Taxol resistance in colorectal cancer. Targeting the YAP-COX2 pathway may be important to inhibiting chemotherapeutic drug resistance in tumors with high YAP-COX2 levels [101].
In 5-FU-resistant colon cancer cells, c-Yes interacts with YAP and activates YAP by promoting its nuclear accumulation. Increased YAP expression was found in 5-FU-resistant colon cancer liver metastases, and high expression of YAP is associated with shorter survival [102]. YAP increases EGFR expression in esophageal carcinoma, causing resistance to 5-FU and docetaxel [103].
Cisplatin is a DNA-damaging agent used to treat many cancers [104]. In cisplatin-resistant oral squamous cancer, p-YAP decreased, whereas nuclear YAP increased and knockdown of YAP increased cisplatin sensitivity [105]. YAP knockdown reduces the DNA damage response in urothelial carcinoma cells and promotes DNA damage. Nuclear YAP expression predicts poor outcomes in urothelial carcinoma patients treated with chemotherapy [106]. The expression levels of YAP and its target genes increased in cisplatin-resistant ovarian cancer cells through an increase in autophagy and autolysosome degradation. Meanwhile, downregulation of YAP sensitizes drug-resistant ovarian cancer cells to cisplatin [107].
Chemotherapy resistance is also associated with the dysregulation of miRNAs, which are upstream regulators of YAP. In Taxol-resistant ovarian cancer cells, miRNA-363 is upregulated and involved in resistance by targeting LATS2, which results in YAP activation [108].
3.2 Targeted therapy
As a receptor tyrosine kinase (RTK), EGFR is frequently mutated or amplified during tumorigenesis. Many EGFR antibodies and small-molecule inhibitors have been developed to treat cancer. YAP activation is associated with cetuximab resistance, an EGFR monoclonal antibody [77], whereas YAP inhibition re-sensitizes colorectal cancer cells to cetuximab [109]. In addition, numerous studies have demonstrated that YAP affects resistance to various small-molecule inhibitors of EGFR [110 –121]. For example, YAP expression is associated with NSCLC resistant to erlotinib, gefitinib, and T790M-specific osimertinib, whereas inhibition or inactivation of YAP results in re-sensitization to these tyrosine kinase inhibitors (TKIs) [116, 119]. Nuclear YAP expression is associated with poor outcomes in lung adenocarcinoma patients treated with EGFR inhibitors [122]. EGFR TKI, together with a YAP inhibitor, prolonged the survival time of lung cancer patients [113]. Downregulating the mechanosensitive transcription of YAP using siRNA or inhibiting YAP with verteporfin (VP), a small-molecule inhibitor of the YAP–TEAD interaction, reversed lapatinib resistance [112]. In HCC, hypoxia and cirrhosis stiffness affects resistance to sorafenib and is associated with the activation of YAP. Inhibition of YAP re-sensitizes HCC to sorafenib [120, 121]. In addition, CTGF and AXL, two YAP-targeted genes, are potential therapeutic targets for overcoming EGFR inhibitor resistance [115].
Many studies have identified mechanisms of YAP-mediated resistance to KRAS signaling inhibition. In response to treatment with a BRAF and MEK inhibitor, YAP induced Bcl-XL expression to block apoptosis caused by BRAF and MEK inhibitors [123]. Depletion of YAP sensitized PDAC cells with a KRAS mutation to a pan-RAF inhibitor by suppressing AKT pathway activation [124]. Inhibition of YAP activity with VP induced the re-sensitization of melanoma cells to a BRAF inhibitor [125]. Another study reported that increased YAP activity increases the expression of E2F1-related cell cycle genes and activates the EGFR, Myc, and AKT signaling pathways, resulting in the BRAF inhibitor resistance in melanoma cells. However, down-regulation of YAP suppresses the resistance phenotype [126].
Abnormal PI3K/mTOR signaling pathway activation may occur in many tumors. In ovarian cancer, the expression of YAP has been associated with resistance to a PI3K/mTOR inhibitor [127]. Downregulation of YAP sensitizes ovarian cancer cells to AZD1775, a small-molecule WEE1 kinase inhibitor. YAP inactivation enhanced DNA damage and mitotic failure caused by AZD1775 [128]. Inhibition of the SOX2 transcription and YAP activation increased sensitivity to imatinib mesylate [129]. In ER+ breast cancer, resistance to CDK4/6 inhibitors is associated with FAT1 genomic loss and CDK6 upregulation mediated by YAP activity [130].
3.3 Radiotherapy
Radiotherapy is considered a modality of choice for many cancers. It causes DNA strand breaks, cell cycle exit, and apoptosis. In head and neck squamous cell carcinoma, high levels of YAP predict poor response to radiotherapy [131]. YAP knockdown in urothelial carcinoma cells promotes the DNA damage response induced by radiation exposure [106]. In medulloblastoma, YAP overexpression promotes the survival of cells following irradiation. Downregulation of YAP reduces the radiation dose required to induce cell death [132]. YAP causes cells to enter mitosis with unrepaired DNA by inducing IGF2 expression and AKT activation, which leads to ATM/Chk2 inactivation and escape from cell cycle checkpoints [132]. We found that high levels of YAP are associated with poor prognosis in glioma patients treated with radiotherapy. Radiation drives YAP into the nucleus, followed by activation, which drives glioma cell growth by promoting the expression of FGF2 and subsequently activating the MAPK signaling pathway. Activation of the YAP-FGF2-MAPK pathway contributes to the enhancement of DNA repair, promoting cell cycle progression and inhibition of apoptosis, resulting in glioma cell survival following radiation. Blocking YAP-FGF2-MAPK by AZD4547 sensitizes gliomas to radiotherapy [57].
3.4 Immunotherapy
Immunotherapy is currently an active area of cancer research. The immune checkpoint receptor PD-1 and ligand PD-L1 function in the main immune checkpoint pathway, inducing T cell exhaustion and preventing cytotoxic T cell-mediated tumor killing. Numerous studies have indicated that YAP activation promotes the expression of PD-L1 in many tumor types, such as breast cancer, NSCLC, mesothelioma, and melanoma [133 –137], suggesting that PD-L1 is a direct transcriptional target of YAP. YAP upregulates PD-L1 expression in BRAF inhibitor-resistant melanoma, which mediates escape from cytotoxic T cells [135]. Furthermore, analysis of TCGA database showed a strong correlation between YAP and PD-L1 expression in lung adenocarcinoma [136].
MDSC and tumor-associated macrophages (TAM) are myeloid immune cells that effectively inhibit T cells’ function and secrete cytokines to promote tumor development. In a Pten−/−Smad4−/− prostate cancer model, YAP promotes the recruitment of MDSCs by increasing the expression of CXCL5, which attracts CXCR2-expressing MDSCs to the tumor microenvironment [138]. YAP also recruits MDSCs by inducing the expression of IL-6, CSF1-3, TNFα, IL-3, CXCL1/2, and CCL2 in PDAC and breast cancer stem cells, whereas YAP knockout promotes macrophage reprogramming and reactivation of the immune response [86, 139]. Activation of YAP results in TAM recruitment by inducing CCL2 and CSF1 expression, which promotes tumor development [140, 141]. YAP also functions in the accumulation of regulatory T cells (Tregs), which are enriched in many tumors and associated with suppressing antitumor immunity. Downregulation of YAP significantly inhibits the immunosuppressive effect of Tregs [142]. Tumors exhibiting YAP overexpression are resistant to the immunotherapy agents, Pembrolizumab and MLN8237 [143, 144].
4 YAP-targeting drugs
Since YAP functions in tumorigenesis and anticancer drug resistance, targeting YAP may benefit patients. The most widely used strategy for YAP inhibition is to block the combination of YAP and TEAD. Here, we introduce three drugs and their mechanisms in Fig. 3.
VP, a benzoporphyrin derivative containing an aromatic heterocyclic ring molecule, is composed of four modified pyrrole units bonded to a carbon atom through a methane bridge [145, 146]. As an FDA-approved drug for treating wet aged-macular degeneration, VP acts as a photosensitizer and exhibits its primary biological activity through the action of short-lived single oxygen and reactive oxygen radicals. VP is capable of binding to YAP and disrupting the YAP-TEAD complex [147]. Treatment with VP in cancer cells inhibits the expression of YAP target genes, such as CYR61, CTGF, AXL, and BIRC5 [148, 149]. In the present study, we found that VP can enhance the sensitivity of radiotherapy in gliomas by inhibiting the activity of YAP [57].
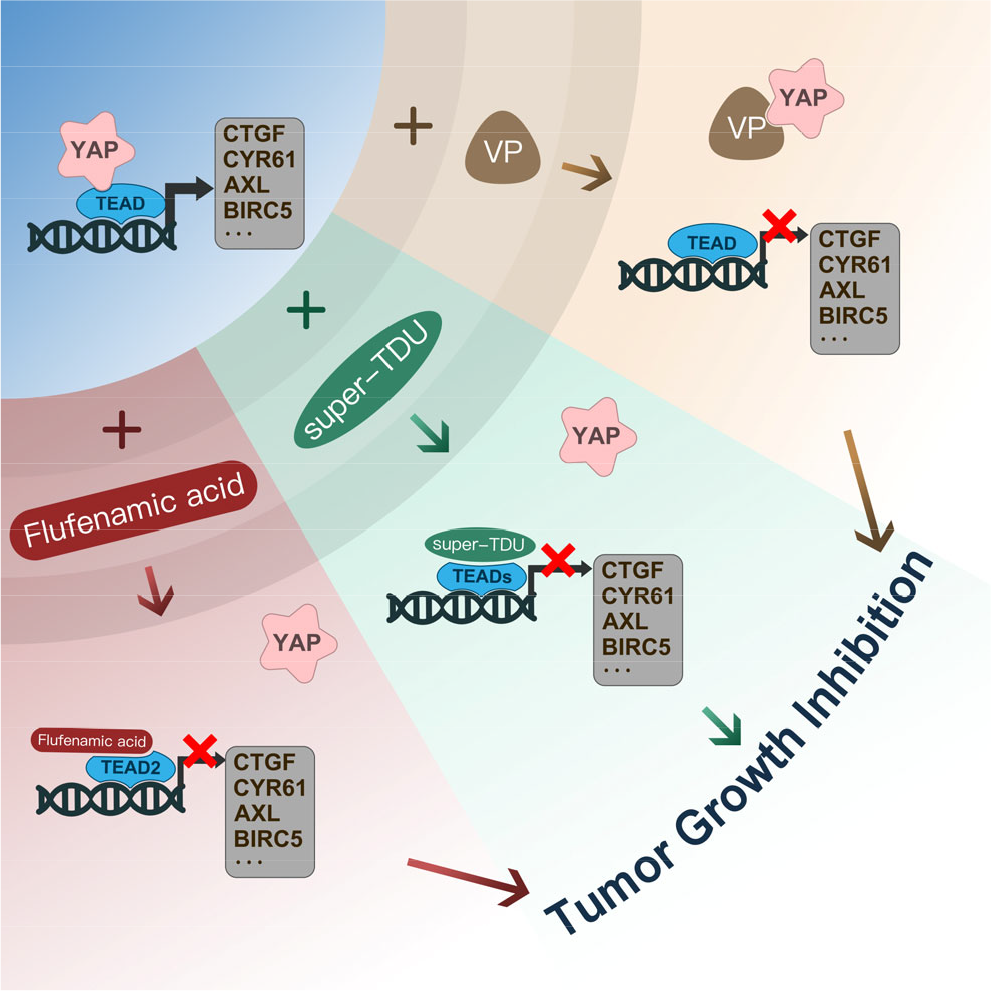
YAP inhibitors target the YAP–TEAD interaction. Three drugs reduced the activity of YAP by preventing the formation of the YAP-TEAD complex.
As a tumor suppressor, VGLL4 competes with YAP for binding to TEAD and, therefore, inhibits EMT and cancer progression [150, 151]. Because VGLL4 binds to TEAD4 via the TDU domain located at its C-terminus, a peptide mimicking VGLL4, known as super-TDU, was developed to block the YAP–TEADs interaction [152]. In gastric cancer, super-TDU blocks YAP activity at the transcriptional level, which offer a strategy for cancer treatment [152].
Flufenamic acid, a nonsteroidal anti-inflammatory drug, binds to the central pocket of TEAD2 and inhibits YAP activity [153]. Another study found that flufenamic acid binds to the conserved cysteine in the lipid pocket, thus inhibiting the YAP–TEAD interaction, transcriptional activity, and glioblastoma growth in vitro [154].
Toxicity is a significant challenge to successfully implementing YAP inhibitors in the clinic. Depletion of YAP in the liver [155], heart [156], and lung [157] may induce organ failure and hypoplasia. Additionally, renal toxicity was observed in mice administered K-975, which disrupts the YAP–TEAD interaction and exhibits anti-tumorigenic properties in malignant pleural mesothelioma cells [23]. Moreover, while YAP is highly expressed across most solid tumors, it is silent in blood malignancies [158, 159]. Further studies are needed to fully explore the effects of targeting YAP in the circulatory system. The ideal dose must be determined by a large number of experiments to minimize side effects. It is important to identify a therapeutic dose and time window for these inhibitors to safely administer the drug to improve patient survival without causing toxicity.
Although several drugs directly target YAP, as mentioned in this review and presented in Fig. 3, to our knowledge, none have advanced to the clinical stage. Other compounds reported to target YAP indirectly have been reported elsewhere [160]. YAP is not an enzyme, so it is more difficult to target with small-molecule drugs. Therefore, there is an urgent need to identify new inhibitors. Macromolecular drugs generally act on cell surface targets and inhibit protein-protein interactions. Despite their strong specificity, these drugs cannot be taken orally. Small molecules can be taken orally and can readily cross the cell membrane to act on intracellular targets. It is difficult for small molecules to act directly on proteins. In the future, drugs need to be modified and improved to achieve better inhibitors with low toxicity and high efficiency.
5 Conclusion
In the last decade, growing evidence indicates that the Hippo pathway plays a significant role in cancer progression. YAP exhibits antitumor drug resistance as a key protein in the Hippo pathway. Inhibiting YAP activity in drug-resistant cancer cells can sensitize them to anticancer therapy in various resistance models. Therefore, identifying YAP inhibitors will provide a new option for cancer treatment. FDA-approved non-anti-cancer drugs, such as VP, are under clinical trials to assess their effectiveness against cancer. In the future, more efforts should be made to identify more effective YAP inhibitors, including blockers of the YAP–TEAD interaction or drugs that directly target YAP. We look forward to developing more anti-YAP compounds to be tested as new anticancer treatments. In addition, the molecular basis of anticancer therapy resistance has not been fully elucidated. Identifying the underlying mechanism needs will be valuable for designing new anticancer strategies. Furthermore, the pathogenesis of malignant tumors is complex and it is difficult to suppress only one signaling pathway to eradicate tumor. A combination of anti-YAP therapies with other signaling pathway inhibitors may yield synergistic effects against cancer.
Footnotes
Conflict of interests
The authors declare that they have no competing interests.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81872053, 81902526, 82072770) and the Natural Science Foundation of Jiangsu Province (Grant No. BK20201458).
Authors’ contribution
Yu Zhang: Conceptualization (lead); data curation (lead); writing-original draft (lead). Xiang Wang: Writing-original draft (supporting). Xiu-Ping Zhou: Conceptualization (lead); supervision (lead); writing-review & editing (lead).