
Abstract
Sleep constitutes a third of human life and it is increasingly recognized as important for health. Over the past several decades, numerous genes have been identified to be involved in sleep regulation in animal models, but most of these genes when disturbed impair not only sleep but also health and physiological functions. Human natural short sleepers are individuals with lifelong short sleep and no obvious adverse outcomes associated with the lack of sleep. These traits appear to be heritable, and thus characterization of the genetic basis of natural short sleep provides an opportunity to study not only the genetic mechanism of human sleep but also the relationship between sleep and physiological function. This review focuses on the current understanding of mutations associated with the natural short sleep trait and the mechanisms by which they contribute to this trait.
1 Introduction
We recognize the importance of sleep from everyday experience, but why we need to sleep remains an enigma. Vast literature has shown that sleep is crucial for cardiovascular functions [1], metabolic homeostasis [2], immune reactions [3], mental state [4] and cognition [5]. Increasing evidence indicates that both too much and too little sleep are associated with adverse health conditions [6]. Therefore, appropriate quantity of daily sleep is critical for health and normal physiological functions.
Genetic screens have identified many genes that affect sleep in different organisms from worms to rodents, indicating that the molecular mechanisms of sleep regulation are evolutionarily conserved [7]. However, most (if not all) of these mutations that have been tested result in impaired physiological function and organismal fitness, such as deficits in learning and memory and reduced life span [8 –12]. In contrast, human natural short sleepers are individuals who sleep significantly shorter than the average population without a desire for more sleep and without any obvious negative health consequences [13]. These individuals feel well although they sleep only 4–6.5 h per day, which is not observed in average sleepers. In addition, they have more flexible timings of sleep and report no jet lag. Given that the natural short sleep (NSS) trait is inheritable, dissecting its genetic basis provides a unique opportunity to study the molecular pathways underlying the need for sleep. Here, we provide an overview of genetic mutations identified to be associated with NSS and how they lead to reduced sleep.
2 DEC2
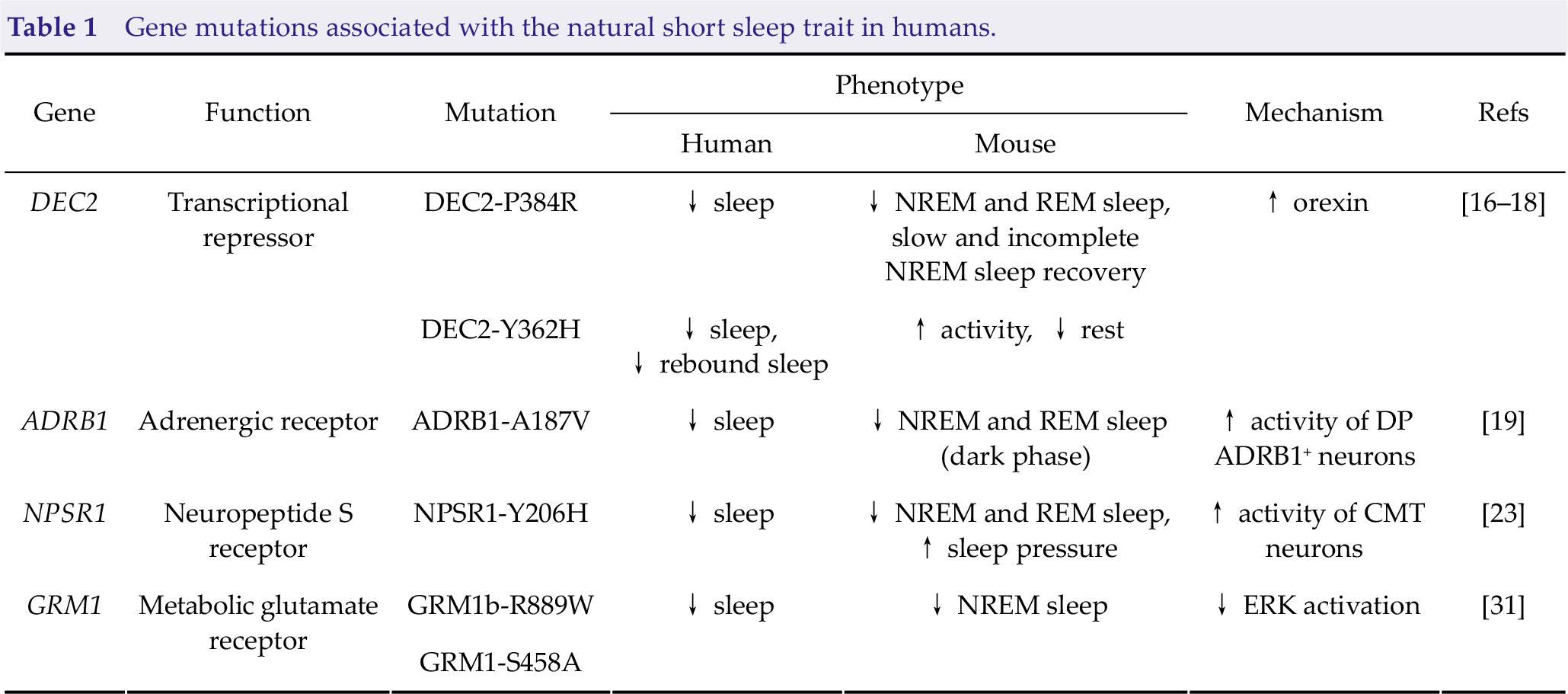
The first gene reported to be associated with NSS is DEC2, which encodes a transcriptional repressor that belongs to the basic helix-loop-helix subfamily [14]. It is a negative component of the circadian clock and represses CLOCK/BMAL1-induced transactivation through E-box elements [15]. In a small family, He et al. [16] identified a rare point mutation in DEC2 via candidate gene sequencing and found that heterozygous carriers of this mutation had lifelong shorter daily sleep duration (on average 6.25 h) compared to normal individuals (on average 8.06 h). This mutation results in a proline-to-arginine substitution at amino acid residue 384 of DEC2 (DEC2-P384R), which is a site conserved in mammals. To confirm whether the short sleep phenotype was indeed caused by this mutation, He et al. generated transgenic mice using a human bacterial artificial chromosome clone carrying wild-type (WT) or mutant DEC2. DEC2-P384R mice demonstrated a longer activity period with significantly reduced non-rapid eye movement (NREM) sleep and rapid eye movement (REM) sleep duration during the light phase (rest phase), but there was no significant difference in NREM delta or REM theta power. Analysis of sleep architecture revealed an increased NREM sleep bout number and decreased bout duration in DEC2-P384R mice, suggesting more fragmented sleep. Moreover, DEC2-P384R mice displayed slower and incomplete NREM sleep recovery after 6 h of sleep deprivation compared with WT mice. The successful recapitulation of the short sleep phenotype in mouse models strongly suggests that the DEC2-P384R mutation leads to NSS in humans, likely by impinging on sleep homeostasis regulation. Further evidence for a role of DEC2 in sleep regulation comes from a study [17] reporting the DEC2-Y362H mutation to be associated with reduced sleep duration and less recovery sleep after sleep deprivation in humans. Notably, in a pair of twins, the DEC2-Y362H carrier had considerably shorter total sleep duration (299.3 min) than his non-carrier brother (364.7 min). The carrier also exhibited less recovery sleep following sleep deprivation and fewer performance lapses during sleep deprivation.
How do these DEC2 mutations shorten sleep duration? Hirano et al. [18] found that the actions of mutant DEC2 may be mediated by the wake-promoting peptide orexin. Orexin is known to be generated from a neuropeptide precursor protein encoded by the hypocretin (Hcrt) gene, which displayed upregulated expression in the hypothalamus of DEC2-P384R and DEC2-Y362H mice compared with DEC2-WT mice. Further molecular characterization demonstrated that DEC2 binds to the E-box sequence (CAGCTG) in the promoter region of Hcrt and suppresses its expression. However, the mutant DEC2 showed impaired repressor activity relative to WT DEC2, resulting in increased Hcrt expression. In addition, the administration of a nonselective orexin receptor antagonist (MK-6096) could partially rescue the short sleep phenotype in DEC2-P384R mice. Taken together, these findings suggest that DEC2 mutations reduce sleep by enhancing orexin production.
3 ADRB1
Ten years after the first NSS-related mutation was reported, ADRB1, which encodes the β1-adrenergic receptor, emerged as the second gene associated with NSS [19]. The β1-adrenergic receptor, a receptor activated by noradrenaline and adrenaline, plays an important role in regulating the activity of the sympathetic nervous system [20]. Within the brain, noradrenaline—mainly generated by the locus coeruleus—promotes arousal by activating α1- and β-adrenergic receptors in the medial septal and medial preoptic areas [21]. Shi et al. [19] identified a rare mutation in ADRB1 (ADRB1-A187V) in a family with natural short sleepers via whole-exome sequencing. This A187 residue is located in the fourth transmembrane domain of ADRB1 and is highly conserved from invertebrates to humans. Heterozygous mutation carriers slept 5.7 h per day on average, which was much shorter than the sleep duration of the non-mutation carriers (7.9 h). To validate that this is indeed a causative mutation for NSS, Shi and colleagues generated Adrb1-A187V knock-in mice using the CRISPR-Cas9 strategy. Heterozygous mutants showed significantly reduced sleep duration during the dark (active) phase, strongly suggesting that the ADRB1-A187V mutation contributes to NSS in human carriers.
To explore the mechanism by which ADRB1-A187V leads to shortened sleep, Shi et al. [19] first demonstrated that this mutation resulted in decreased stability of the ADRB1 protein and thus decreased protein levels. Next, by conducting calcium imaging, they found that dorsal pons (DP) ADRB1+ neurons were active during wakefulness and REM sleep while remaining quiescent during NREM sleep. Moreover, optogenetic activation of DP ADRB1+ neurons during NREM sleep elicited immediate NREM-to-wake transitions, indicating that these cells are primarily wake promoting. In the mutant mice, DP ADRB1+ neuronal activity was enhanced, which was associated with increased wakefulness and decreased sleep duration. These findings indicate that ADRB1 is involved in sleep regulation, and the A187V mutation further enhances the activity of the wake-promoting ADRB1+ neurons, which in turn results in reduced sleep. As a well-studied attractive drug target [22], ADRB1’s role in sleep regulation may render it a potential therapeutic target for the treatment of sleep disorders.
4 NPSR1
In 2019, Xing et al. [23] reported another NSS-associated mutation located in the NPSR1 gene, which encodes neuropeptide S receptor 1. NPSR1 is a G-protein-coupled receptor, and its cognate ligand, neuropeptide S (NPS), has been reported to activate histaminergic and orexinergic neurons in the posterior hypothalamus to promote wakefulness [24, 25]. NPS has also been shown to exert wake-promoting effects by depolarizing GABAergic neurons in the ventrolateral preoptic nucleus [26]. Using whole-exome sequencing, Xing et al. identified a rare point mutation in NPSR1 in a small family with natural short sleepers [23]. This mutation causes a tyrosine to a histidine substitution at residue 206 (Y206H), and the two heterozygous carriers slept only 5.5 and 4.3 h per day, respectively. To confirm that the NPSR1-Y206H mutation was causal for the short sleep trait in the carriers, Xing et al. generated Npsr1-Y206H knock-in mice using a CRISPR-Cas9 approach and assessed sleep in these animals. Heterozygous mutant mice exhibited reduced NREM and REM sleep, particularly in the dark phase, supporting the notion that the mutation results in NSS in human individuals.
NPSR1 mRNA is highly expressed in the centromedial thalamus (CMT) [27], a region that modulates brain-wide cortical activity during sleep and provides dual control of sleep-wake states [28]. Optogenetic activation of CMT neurons reliably induces rapid awakening from NREM sleep. Moreover, activation of CMT neurons promotes sleep recovery following a 4 h sleep deprivation and silencing them delays this process. CMT neurons in Npsr1-Y206H mice showed enhanced response to NPS treatment. In addition, the cAMP response element-binding protein (CREB) is believed to be a downstream effector of NPSR1 signaling [29], and NPS induces an increase in CREB phosphorylation in the cortex [23]. This increase in phospho-CREB levels elicited by NPS is also enhanced in the mutants. This series of results suggests that the Y206H mutation increases NPSR1 signaling and hyperactivates CMT neurons, leading to shortened sleep duration.
To investigate whether Npsr1-Y206H affects cognitive performance, Xing et al. measured contextual memory in mutant and control mice using the contextual fear conditioning test [23, 30]. After training, Npsr1-Y206H mice exhibited performance comparable to WT mice, indicating that learning and memory were intact in the mutants. However, WT mice that underwent 6 h sleep deprivation immediately after training showed impaired memory, whereas mutant mice did not, which suggests that contextual memory was more resistant to sleep loss in Npsr1-Y206H mice. Overall, Npsr1-Y206H shortened sleep duration without impairing learning and memory in mice, which appears to be consistent with what has been observed in human carriers.
5 GRM1
In a recent study of two unrelated families with natural short sleepers [31], whole-exome sequencing unveiled two independent rare mutations in the GRM1 gene (GRM1b-R889W and GRM1-S458A), which encods glutamate metabotropic receptor 1. Group I metabotropic glutamate receptors (mGluRs 1 and 5) are family C G-protein-coupled receptors participating in the modulation of synaptic transmission and neuronal excitability [32]. Previous studies reported mGluR5 to be involved in sleep regulation [33, 34], whereas it was unknown whether mGluR1 was also involved. In one of the families with natural short sleepers, three heterozygous carriers of GRM1b-R889W mutation slept 6.05, 5.15, and 5.10 h per day, respectively, which was considerably less than the non-affected family members (on average 7.32 h) [31]. In the other family, a heterozygous GRM1-S458A carrier slept 6.00 h per day, and his non-carrier sister slept 6.93 h per day.
Amino acid sequence analysis revealed that the two mutations are located in different domains, with GRM1b-R889W in the C-terminal intracellular domain of the mGluR1b isoform and GRM1-S458A in the large N-terminal extracellular domain [31]. Extracellular signal-regulated kinase (ERK) is known to be activated by mGluR [35], and the expression of both GRM1b-R889W and GRM1-S458A resulted in reduced ERK phosphorylation in cell culture, indicative of reduced activation. A previous study demonstrated that the ERK signaling pathway regulates sleep duration and inhibition of ERK phosphorylation decreases sleep in mice [36], consistent with the effects of these GRM1 mutations.
6 Discussion and conclusion
Perhaps the most intriguing question regarding the NSS trait is how these individuals maintain such short sleep duration lifelong with no obvious health or physiological consequences. The simplest explanation is that these individuals have an altered sleep homeostasis and thus need less sleep. However, among these four genes (Table 1), only the DEC2 mutation appears to result in reduced recovery sleep following sleep deprivation in mice [16], indicating decreased sleep need. Neither Npsr1 nor Gmr1 mutations significantly alter sleep rebound in mutant mice, but this aspect has not been assessed in Adrb1 mutant mice [19, 23, 31]. Instead of impinging on sleep homeostasis, most of these mutations (DEC2, ADRB1, and NPSR1) lead to enhanced wake-promoting signals, suggesting that the nature of NSS is actually increased wake period rather than reduced sleep. This increased wake period may not trigger accumulation of sleep debt, and thus natural short sleepers do not appear to experience the adverse outcomes of sleep deprivation. Electroencephalography power analysis and sleep deprivation experiments conducted in human natural short sleepers are needed to address this issue.
Gene mutations associated with the natural short sleep trait in humans.
Another important question is whether the natural short sleepers are “completely normal and healthy”. Although these individuals do not display any obvious physical or mental conditions associated with the lack of sleep, there may exist certain some adverse effects that have not yet been discovered. Animal studies to date have focused mostly on sleep analysis, and systematic assessment of other physiological indices and behaviors will need to be performed to answer this question. Apart from genetic factors, environmental factors also exert major influences on sleep [37 –42]. All animal studies to date have been conducted under baseline conditions, whereas humans experience many stressful challenges throughout their lifetime. This discrepancy may also account for the relatively weaker sleep phenotype in mutant mice than in the human mutation carriers in some cases. It would be interesting to assess how NSS-associated mutations alter sleep and other physiological functions or behaviors under conditions such as starvation, infection, and social isolation.
Overall, our current understanding of the molecular mechanism underlying NSS is quite limited. More systematic and in-depth investigations need to be conducted to unveil the intricate relationships between sleep, wakefulness, and other physiological functions in natural short sleepers. Identifying why natural short sleepers do not need to sleep as long as normal individuals may improve the understanding of the need for sleep.
Footnotes
Conflict of interests
All contributing authors report no conflict of interests in this work.
Funding
This work was supported by grants from the Ministry of Science and Technology of China (Grant No. 2021ZD0203202), and the Natural Science Foundation of China (Grant Nos. 31930021 and 32022035).