
Abstract
Gliomas, the most common primary tumors in the central nervous system (CNS), can be categorized into 4 grades according to the World Health Organization. The most malignant glioma type is grade Ⅳ, also named glioblastoma multiforme (GBM). However, the standard treatment of concurrent temozolomide (TMZ) chemotherapy and radiotherapy after maximum resection does not improve overall survival in patients with GBM. Targeting components of the CNS microenvironment represents a new strategy for improving the efficacy of glioma treatment. Most recent studies focused on T cells. However, there is a growing body of evidence that tumor‐associated macrophages (TAMs) play an important role in tumor progression and can be regulated by a wide array of cytokines or chemokines. New TAM‐associated immunotherapies may improve clinical outcomes by blocking tumor progression and prolonging survival. However, understanding the exact roles and possible mechanisms of TAMs in the tumor environment is necessary for developing this promising therapeutic target and identifying potential diagnostic markers for improved prognosis. This review summarizes the possible interactions between TAMs and glioma progression and discusses the potential therapeutic directions for TAM‐associated immunotherapies.
1 Introduction
Gliomas are the most common primary brain tumors. Gliomas affect the central nervous system (CNS) by secreting signaling molecules that influence the microenvironment. Gliomas can be categorized into subtypes based on different methods. Histologically, gliomas can be grouped into astrocytoma, oligodendroglioma, and oligoastrocytoma, and gliomas can be classified into four grades based on malignancy [1]. Of note, the grade of glioma and the malignant degree are positively correlated. Grade Ⅳ gliomas, also called glioblastoma multiforme (GBM), often present the worst prognosis and are a deadly threat for patients. The current treatment for GBM is maximum surgical resection with radiotherapy and temozolomide (TMZ) based chemotherapy. Unfortunately, survival in GBM patients is still less than 15 months after diagnosis [2] because of the high rate of metastasis and recurrence [3]. Hence, exploring new therapeutic approaches for GBM is crucial.
Interestingly, the glioma microenvironment differs from the physiology of the broken blood–brain barrier (BBB), which is induced by pro‐inflammatory molecules produced by microglia and tumor cells [2]. Tumor‐associated macrophages (TAMs), the most abundant cell population in the microenvironment, consist of resident CNS microglia and peripheral monocytes, which easily infiltrate into the CNS via the damaged BBB [4, 5]. The formation of the fully‐functioning TAMs requires multiple phases, which will be discussed in further detail below. TAMs are recruited to either the primary tumor or metastasis tumor region. Then, TAMs are polarized into M1 or M2 phenotypes, with anti‐tumor and pro‐tumor functions, respectively, by chemokines and cytokines. The chemokines and cytokines are secreted by the malignant cells or other non‐neoplastic cells in the microenvironment [3, 6]. TAMs can exert dual influences on glioma progression, one of which promotes tumor progression and increases metastasis burden in the CNS in a highly lethal manner [7]. Overall, the glioma microenvironment may facilitate malignant tumor progression and recurrence [8]. Therefore, targeting the TAMs in the CNS has the therapeutic potential to improve the prognosis of patients with gliomas.
2 Tumor‐associated macrophages in the glioma tumor microenvironment
2.1 Glioma tumor microenvironment
Under normal physiological conditions, the CNS hosts a unique microenvironmental condition that differs significantly from most other organs and tissues [9]. Under neuropathological conditions, the tumor microenvironment (TME) consists of a variety of non‐neoplastic cells, cytokines, and extracellular matrix in addition to neoplastic cells [10]. The non‐neoplastic cells consist of multiple cell types, including fibroblasts, epithelial cells, granulocytes, mast cells, and macrophages [11]. A majority of non‐neoplastic cells are TAMs, which occupy up to 50% of the tumor mass [12] and are believed to regulate tumor evolution by supporting its expansion or suppressing its progression.
In recent years, a large body of literature has been devoted to the analysis of the TME characteristics after the infiltration of peripheral macrophages. TAMs in gliomas are constantly associated with tumor progression and patient prognosis [13]. The overall TME displays an immunosuppressive status due to the critical permeability of the BBB and less infiltration of lymphocytes [9].
2.2 Origin and recruitment of TAMs
The earliest observation of leukocytes in malignant tumors was in approximately the middle of the 19th century [14]. However, it was not until the 20th century that the specific interaction between TAMs and malignant tumor cells was given attention. Now, there is a growing body of evidence that TAMs play a significant role in forming a suitable TME for tumor growth, progression, metastasis [15, 16].
Under homeostatic conditions, microglia, originating from yolk sac progenitor cells in the early embryo [17], are the main population of brain macrophages, accounting for about 10% of the adult brain cell population [18]. The interaction between these resident microglia and the BBB contributes [19] to the effective protection of the CNS and the maintenance of the immune homeostasis. Under neuropathologic conditions, such as cancer or inflammation, additional peripheral macrophages are recruited to the brain from the bone marrow [10] due to the impaired BBB [20]. Recruitment signals, including CSF‐1, MCP‐1, CCL2, and HGF, are often produced by the tumor cells themselves (Fig. 1) [21 –23]. Inflammatory monocytes, which are precursors of the macrophages, drastically increase in the blood and are recruited to the tumor sites [24]. Interestingly, the peripheral infiltrating macrophages, once activated, are hard to distinguish from the resident microglia, even though their contribution to the pathological TME and tumor progression may be different [25].
TAMs can abundantly populate most solid tumors and can represent up to 50% of the glioblastoma mass [12]. Significantly, the number of infiltrating macrophages is higher in GBM than in astrocytomas or anaplastic astrocytomas in patients with primary brain tumors [9, 26], suggesting that the density of TAMs contributes to a higher degree of malignancy in glioma [27]. Overall, the TAMs, consisting of resident microglia and infiltrating macrophages, occupy a great deal of the microenvironment in gliomas [28] and have the capacity to modify clinical outcomes [29].
2.3 Activation and polarization of TAMs
In addition to recruitment signals, the TME contains a variety of different activation and polarization signals. Mobility and diversity are hallmarks of TAMs. To satisfy the greater demand to control the damage in the CNS, during tumor development or infection, TAMs can be induced into two different subtypes in response to different stimuli [30]. The classically activated macrophages (type‐1 macrophages, M1) are pro‐inflammatory. The alternatively activated macrophages (type‐2 macrophages, M2) are anti‐inflammatory. TAMs are versatile cells that can be polarized in response to different stimuli provided either in vitro [31] or in vivo [32].
Different TAMs, like microglia and macrophages, or M1 and M2 phenotypes, have different responses to TME activation signals. The M1 phenotype responds to Toll‐like receptor 4 (TLR4) ligands and interferon (IFN)‐γ [30], while the M2 phenotype appears after being exposed to anti‐inflammatory molecules, including IL‐4, IL‐10, IL‐13, and glucocorticoids (GCs) [33]. M2 macrophages can be sub- classified into M2a, M2b, and M2c states (Fig. 1) [34].
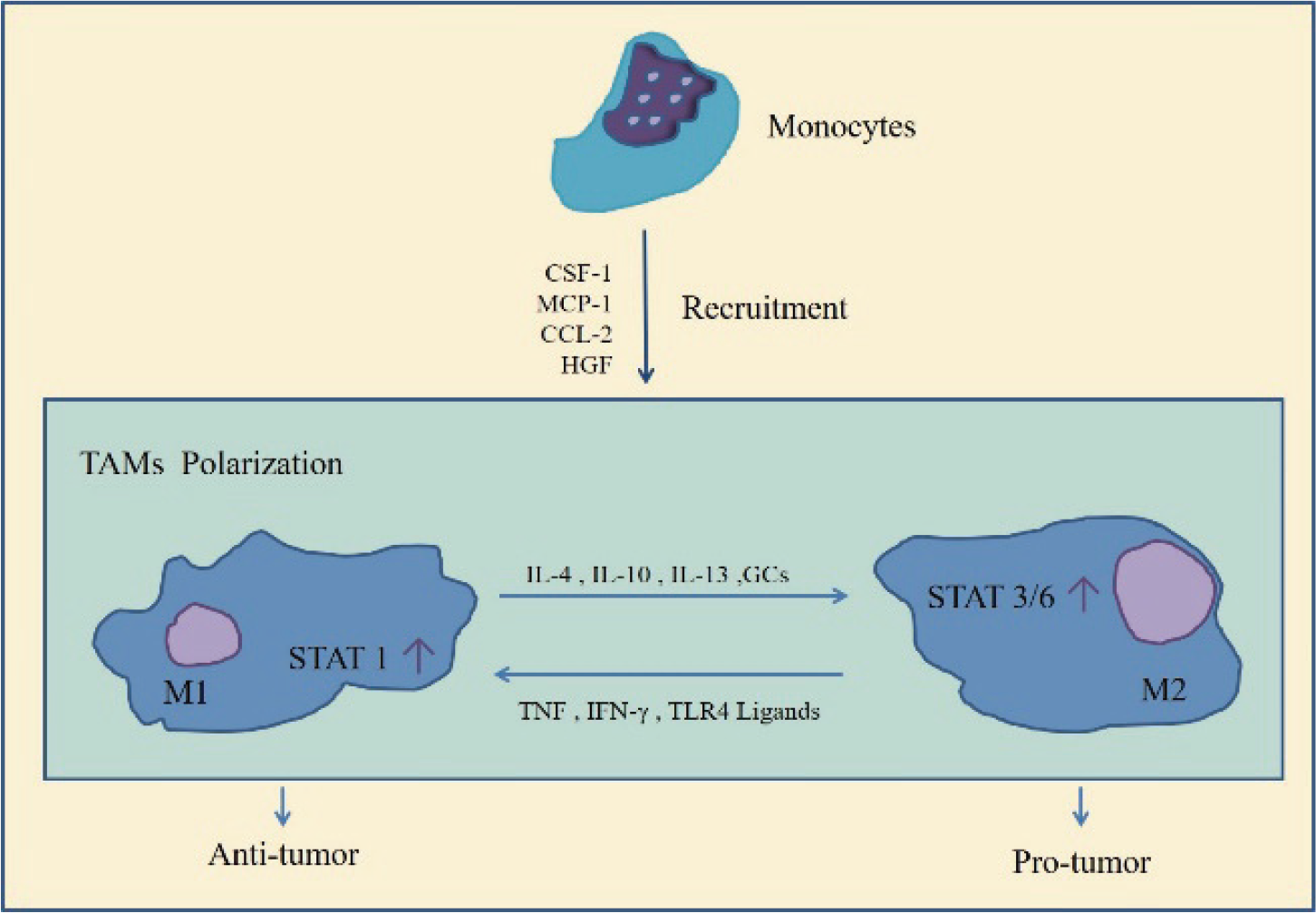
The recruitment and polarization process of tumor‐associated macrophages (TAMs). After being recruited to the central nervous system, TAMs can be polarized into M1 or M2 phenotypes. The M1 and M2 macrophage phenotypes are not fixed. The phenotypes can be converted from one to the other in response to different cytokines in the tumor microenvironment.
Notably, TAMs can alter clinical outcomes by releasing a wide array of cytokines and growth factors, such as vascular endothelial growth factor (VEGF), IL‐8, and basic fibroblast growth factor, in response to factors produced by malignant cells themselves [15]. In this manner, TAMs can either facilitate or suppress tumor growth, proliferation, and metastasis. However, due to the lack of specific markers, identification of M1 and M2 phenotypes in human gliomas is difficult [20]. In addition, in a human glioma single‐cell RNA‐seq‐based transcriptomic analysis, a subpopulation of TAMs was shown to co‐express with M1 (TNF α) and M2 (IL‐10) markers [35].
M2 macrophages are closely related to the growth and progression of gliomas and execute distinct functions that differ from the M1 phenotype. The ratio of M2 macrophages in TAMs correlates with the histological type [36]. In benign tumors, TAMs are mainly M1 type macrophages that can strongly threaten the microorganisms and engulf tumor cells in the TME. In malignant tumors, TAMs are mainly M2 type macrophages that can secrete immunosuppressive factors to promote tumor invasiveness [37, 38]. Taking advantage of the different effects of M1 versus M2 macrophages on tumor progression has emerged as one of the most promising therapeutic options, which we will be explained in Section 4.
3 Role of TAMs in the development of gliomas
M2 macrophages [36] play a pivotal role in immune suppression and pro‐tumor effects during tumor progression. Therefore, the presence of M2 macrophages results in poor clinical prognosis [39]. The capacity of TAMs to facilitate tumor progression relies on promoting microenvironmental immunosuppression, inducing tumor vascularization, and potentiating tumor growth. Overall, there is a growing appreciation that M2 macrophages, which are abundant in primary malignant tumor masses, possess the ability to remodel the microenvironment to support tumor growth. Thus, the main effect of TAMs in gliomas is to promote tumor progression [37].
3.1 TAMs promote immune suppression
Macrophages, either resident microglial cells or infiltrating peripheral macrophages, are a crucial participant in the CNS immune response. TAMs achieve immunosuppression in the glioma TME by generating a great number of immunosuppressive cytokines and chemokines [40]. For instance, interleukin‐10 (IL‐10) plays an essential role in the innate and adaptive immune response and supports the immunosuppressive microenvironment. However, the specific mechanism by which IL‐10 mediates this immunosuppression is unclear. According to a recent study, IL‐10 may promote immune evasion by up‐regulating KPNA2. Tumor progression was significantly reduced after the genetic knockdown of KPNA2 [41]. Overexpression of the indoleamine 2, 3‐dioxygenase (IDO) enzyme also has immunosuppressive potential. IDO can consume tryptophan (Trp) by converting it into kynurenine (Kyn), resulting in immune suppression in the microenvironment [42]. Altogether, the release of IL‐10 and the abundant IDO lead to decreased recruitment and activation of T cells, decreased major histocompatibility complex (MHC) expression, and conversion of M1 to M2 macrophages [43]. Of note, not only are sufficient functional T cells crucial for a powerful T cell immune response, but the capacity to present and recognize antigens is also of great importance. Antigen recognition relies on the co‐expression of human MHC, especially MHC class I (MHC‐1) [44]. Low expression of MHC‐1 promotes resistance to T‐cell‐mediated anti‐tumor effects in the microenvironment [43].
Interestingly, in addition to the exhaustion of T cells, the regulatory T cells (Tregs), which are believed to have the ability to suppress the immune response as well, are increased [45]. Due to the over‐consumption of oxygen caused by tumor growth and abnormal neovascularization, hypoxia develops in the glioma TME. Hypoxia exacerbates immunosuppression by inducing the migration of Tregs to the hypoxic area [43]. One possible mechanism for the migration of Tregs is the hypoxia‐inducible factor 1α (HIF‐1α), which can shunt glucose away from the mitochondria. HIF‐1α induces Tregs to use pyruvate for lipid metabolism under hypoxic conditions. Notably, mild‐type Tregs are more susceptible to inhibition of lipid oxidation than HIF‐1α‐deficient Tregs [46].
3.2 TAMs induce glioma vascularization
Tumor progression relies on angiogenesis. The tumor can have sufficient nutrients and oxygen via diffusion from the TME without any new vessels, if the tumor mass is small [47]. A tumor will not grow, let alone metastasize, beyond 1–2 cm3 size without vascularization to get enough nutrients and oxygen [48]. Once the TAMs are recruited to the tumor region and fully activated, they can produce a wide array of cytokines, chemokines, and growth factors, like VEGF. VEGF is a major contributor to angiogenesis in gliomas. Interestingly, VEGF not only facilitates vascularization and accelerates tumor progression [47, 49], but also contributes to immunosuppression in the glioma TME by blocking the activation of dendritic cells and recruiting Tregs [50].
Apart from VEGF, various signaling molecules also give rise to angiogenesis. In fact, due to resistance in VEGF‐targeted therapy [51], controlling the overexpression of IL‐6 and inhibiting the CXCL2‐CXCR2 signaling pathway have emerged as important therapies in blocking angiogenesis of glioma. IL‐6 is now appreciated as a critical cytokine involved in abnormal angiogenesis. IL‐6 is generated by TAMs and is abundant in the glioma TME [52]. Notably, IL‐6 can promote angiogenesis and facilitate tumor evasion in multiple human cancers [53]. The overexpression of CXCL2 has been observed when isolating fresh TAMs from gliomas. CXCL2 is believed to contribute to vessel formation more than VEGF. Suppression of the CXCL2‐CXCR2 signaling pathway depletes up to 50% of the vessel density and reduces the glioma mass, suggesting that CXCL2 is a crucial participant in the abnormal angiogenesis in gliomas [54].
The hypoxic tumor environment seems to accelerate the process of angiogenesis. Due to accumulated progression and massive angiogenesis of gliomas, the microenvironment eventually becomes hypoxic [43]. The properties of tumor cells, such as tumor progression and invasion, can be influenced by hypoxia [55] or even pushed into a more malignant type [56]. For example, under hypoxic conditions, lactic acid is prone to accumulate in the TME, leading to a low pH environment. The low pH stimulates proangiogenic gene expression [48] and VEGF by stimulating the HIF‐1α‐related signaling pathway [50].
3.3 TAMs facilitate glioma growth
The interaction between TAMs and tumor growth depends on several components of the extracellular matrix. One of the most important components is matrix metalloproteases (MMPs). Elevated MMP levels correlate with glioma growth. TAMs generate a variety of MMPs [57], but most attention has been focused on gelatinases (MMP‐2 and MMP‐9), which facilitate tumor growth and worsen the prognosis. The elevation of MMP‐2 and MMP‐9 in the serum of patients with malignant tumors is closely related to metastasis [58] and has a significant negative impact on survival [59].
Periostin (POSTN) promotes tumor growth and poor prognosis in patients with GBM via a miR‐340‐5p‐macrophage feedback loop [60]. Liu et al. demonstrated that repressing miR‐340‐5p expression can regulate POSTN. POSTN influences αVβ3 integrin and contributes to the recruitment of TAMs and polarization toward M2 macrophages. Interestingly, transforming growth factor β‐1 (TGFβ‐1), produced by the M2 macrophages, downregulates the expression of HMGA‐2 in GBM, which in turn affects miR‐340‐5p expression [60].
MMPs can be induced by many cellular cytokines and chemokines in the glioma TME. For example, TGFβ‐2, secreted by TAMs in the microenvironment, can up‐regulate the expression of MMP‐2. C‐C motif ligand 2 (CCL2), also generated by TAMs or patient‐derived glioblastoma stem cells [61], can support the expression of MMP‐9. Interestingly, CCL‐2 can also have an impact on poor prognosis [62]. The serum levels of CCL‐2 are closely associated with the grade of breast cancer. CCL promotes tumor progression and suppresses overall survival through Smad3 and p42/44 MAPKs signaling [63].
In summary, TAMs are remodeled by neoplastic cells in the glioma microenvironment and play a pivotal role in promoting tumor metastasis by enhancing immunosuppression, inducing angiogenesis, and promoting growth. Of note, most of the cytokines and chemokines involved in tumor metastasis are multi‐functional. For instance, the MMP‐9, described as a significant participant of tumor growth, can also contribute to angiogenesis [59], and VEGF contributes to both immunosuppression and angiogenesis [50].
4 Immunotherapy targeting TAMs in the treatment of gliomas
The current standard treatment for gliomas is TMZ‐based chemoradiotherapy after surgical maximum resection. However, the invasive nature of gliomas, especially of GBM, along with its capacity to infiltrate into adjacent normal brain tissue, leads to TMZ resistance [64] and poor overall survival [65]. The anti‐tumor response of the standard treatment for glioma can be improved if immunotherapeutic strategies are concurrently activated. The CNS has been long recognized as an immune- privileged site [66]. However, there is a growing body of evidence demonstrating that during pathological invasion, the CNS resident microglia and infiltrating macrophages actively participate in the immune response by regulating components of the TME as discussed above. Thus, immunotherapy might be of value in relieving the immunosuppression in the glioma TME [8]. In addition, immunotherapy can take advantage of the immune system’s ability to specifically recognize and respond to malignant or non‐malignant cells, while leaving the normal brain tissue intact [67]. Overall, immunotherapy based on inhibiting the infiltration of macrophages or converting M2 macrophages to M1 highlights the possibility of a more promising long‐term survival in the future.
4.1 Repolarized macrophages from the M2 to M1 phenotype as a therapeutic target
As discussed in Section 2, polarized TAMs can be further subdivided into M1 and M2 phenotypes based on their different surface markers and functional characteristics [68]. Of note, M1 and M2 macrophages have distinct effects on tumor progression [69]. The immunosuppressive M2 phenotype makes up the majority of TAMs in gliomas [15], and these cells promote tumor invasion and angiogenesis [70]. In contrast, pro‐inflammatory M1 macrophages have the capacity for antigen presentation and can, therefore, modulate immune responses against neoplastic cells [71, 72].
The glioma TME can have either anti‐tumoral or pro‐tumoral effects based on the proportion of M2 phenotype in TAMs, and therefore, the TME has a direct impact on glioma histological grade [36, 73]. Promoting the polarization of M2 macrophages toward M1 macrophages in gliomas can enhance the anti‐tumor immune response, suppress tumor growth, and reduce tumor metastasis [74]. Correspondingly, M1 and M2 macrophages are novel potential therapeutic targets because of their plasticity and mobility.
Unlike the permanent phenotypic changes in lymphocytes after exposure to cytokines [75], the switch between M1 and M2 macrophages is dynamic and reversible. The feasibility of reversing an M2 phenotype back to an M1 depends on the molecules in the TME. Of note, M1 and M2 activation of TAMs is strictly controlled by a series of signaling pathways and transcriptional and post‐transcriptional regulatory networks. The abnormal activation of these pathways often occurs during the development of brain tumors or injury [76].
The detailed activation and polarization processes of M1 and M2 are described below. M1 macrophages are often stimulated by pro‐inflammatory cytokines, including TNF, IFN‐γ, and TLR4 ligands [77]. M1 macrophages can present antigens and produce a variety of pro‐inflammatory cytokines, such as IL‐12, IL‐23, type I IFN, CXCL1‐3, CXCL‐5, CXCL8‐10, nitric oxide (NO), and reactive oxygen intermediates (ROI) [78]. In contrast, M2 macrophages tend to be stimulated by IL‐4 or IL‐13 and are often observed in non‐infectious conditions [76]. M2 macrophages produce anti‐inflammatory cytokines like IL‐10 and TGF‐β [79].
The macrophage polarization pathways, especially the phosphorylation of STAT1 and STAT3/STAT6, are crucial for controlling tumor progression. Whether the M1 or M2 phenotype is in dominance in the TME depends on which signaling pathway is dominant. Activation of nuclear factor‐κB (NF‐κB) or STAT1 by lipopolysaccharide (LPS) and IFN‐γ, results in polarization toward M1 macrophages. M1 macrophages respond to the tumor, leading to proinflammatory conditions, including tumoricidal effects and tissue‐damage. Conversely, the M2 phenotype dominates the TME when STAT3 or STAT6 is activated by IL‐4 and IL‐10, enhancing immune tolerance and promoting tumor metastasis [53, 76, 80].
Altogether, the M1 and M2 macrophages form a complex CNS tumor microenvironment. Due to the high production of “killing” cytokines, M1 macrophages are involved in the Th1 response and give rise to tumoricidal activity. On the other hand, M2 macrophages are involved in the Th2 response and promote tissue remodeling and immune tolerance, leading to tumor progression [81].
Chlorogenic acid (CHA), one of the major coffee polyphenols, is an ester of caffeic acid with quinic acid. CHA is in a wide variety of fruits and vegetables [82, 83]. In recent years, the anti‐tumor effects of CHA [82] have been discovered. The therapeutic effect of CHA is due to repolarization of M2 macrophages to M1. Plasticity and mobility are hallmarks of TAMs [84], and TAMs can be remodeled by a CHA‐educated TME. CHA therapy for patients with glioma can skew the macrophages in the TME away from the M2 phenotype and toward the M1 phenotype by stimulating STAT1 and repressing the STAT6 signal pathway. Both STAT1 and STAT6 are critical for M1 and M2 phenotype polarization. Increased expression of M1‐related markers, like MHC Ⅱ and CD11c, and reduced expression of M2 markers, including Arg and CD206, can be detected in vivo or in vitro after CHA treatment. Furthermore, CHA plays a role in inhibiting abnormal vessel formation under hypoxic conditions, which is crucial for tumor progression as described previously. CHA can reduce the transcription of HIF‐1α, which is upstream of VEGF, by inhibiting the HIF‐1α/ AKT signaling pathway and blocking the activation of VEGF and angiogenesis in lung cancer cells [85]. Overall, hopefully, the CHA‐treated TME can result in the inhibition of tumor growth and the reduction in tumor weight [76]. CHA‐associated immunotherapy has the potential to improve glioma therapy.
Other ways to switch M2 into M1 macrophages have promise in prolonging survival. Anti‐CD47 treatment can disturb the CD47‐SIRPα axis and/or Fc mediated tumor cell opsonization and shift toward an M1 predominant phenotype in vivo. The increased M1 macrophage ratio induces the glioma TME to exhibit anti‐tumor properties, which enhance neoplastic cell phagocytosis [73, 86, 87].
Inhibition of the colony‐stimulating factor‐1 receptor (CSF‐1R) can depolarize macrophages from M2 macrophages to compromise glioma progression and improve long‐term survival. CSF‐1, along with CSF‐2, stimulates TAM polarization. CSF‐2 promotes pro‐tumor effects, while CSF‐1 inhibits tumor growth, blocks angiogenesis, and prolongs the survival [88]. According to Sun et al., CSF‐1R correlates with the histologic grade of glioma because of its effects on extracellular signal‐regulated kinase 1/2 (ERK1/2) signaling. Disrupting the CSF‐1R can indeed repress tumor migration [89].
Disruption of NF‐κB can also switch the M2 phenotype to M1 macrophages, resulting in tumor regression in the ovarian tumor model [90, 91]. Furthermore, disruption of NF‐κB p50 expression in both TAMs and T cells can lead to a compromise in GBM immune evasion by suppressing M2 polarization. Analyzing the differences in TAMs, T cells, and survival between wild‐type mice and p50 mice with GL261‐Luc glioblastoma cells demonstrated that mice lacking p50 favor an M1 macrophage dominant microenvironment, which contributes to the reduction in tumor mass. Hence, tumor progression is greater in p50 mice than in wild‐type mice, which ultimately shortens overall survival. In summary, NF‐κB p50, in both tumor T cells and TAMs, is a potential target for GBM immunotherapy [92].
Remodeling M2 macrophages toward the M1 phenotype is a new potential strategy for inhibiting angiogenesis and improving survival in patients with gliomas. To verify this treatment, Cui et al. built an organic brain tumor microenvironment in vitro composed of biomimetic cells, inflammation‐related cytokines, and extracellular matrix and conducted a series of experiments. Blocking the transforming growth family β‐receptor 1 (TGFβ‐R1) and the αβ can significantly control the neovascularization when M1‐like macrophages switch toward M1 macrophages, relieving the immune suppression and prolonging survival [93].
Overall, regulating the glioma microenvironment or directly targeting the signaling pathways involved in TAM polarization toward the M1 phenotype shows promising potential in relieving the immunosuppression, arresting the proangiogenic behaviors, and improving the long‐term survival in patients with gliomas.
4.2 Inhibiting TAM recruitment as a therapeutic target
A variety of immunotherapies targeting macrophage recruitment have emerged. Instead of directly targeting TAMs, regulating the glioma TME to interfere with the recruitment of TAMs also demonstrates the potential for improving treatment of gliomas. POSTN, produced by glioma stem cells (GSCs), contributes to the density of TAMs in the glioma TME and, therefore, promotes tumor growth [60]. In line with this, down‐regulating the expression of POSTN leads to the inhibition of TAM recruitment through the integrin αvβ3 signaling pathway. Decreased TAMs prolongs the survival of mice with GSC‐derived xenografts [94].
In addition, the inhibition of CXC chemokine receptor 2 (CXCR2) can halt tumor progression when applied at the beginning of tumor growth in vivo. CXCL2, a member of the CXC family, plays a significant role in tumor progression by promoting TAM recruitment and tumor angiogenesis. Thus, CXCL2 is a new therapeutic target for treating glioma. The CXCL2 receptor, CXCR2, is closely related to glioma malignant grade and tumor recurrence. The interaction between CXCL2 and G‐protein‐coupled CXCR2 is potentially responsible for partial tumor progression. Inhibition of CXCR2 led to decreased TAM recruitment and vessel density, suggesting a novel opportunity for improving the immunotherapeutic effect [95].
4.3 Treatment combined with concurrent macrophage‐associated immunotherapy
The CNS immune system, which can specifically harm pathological cells rather than healthy cells, is tightly related at multiple immune checkpoints to prevent excessive immune activation. Tumors can take full advantage of these immune checkpoints to exhibit immune escape [96].
The programmed death‐1 (PD‐1) receptor is the most studied immune checkpoint protein. PD1 inhibition has an outstanding ability to reduce tumor progression and metastasis [97]. PD‐1 is one of the CD28 signaling receptors and exists in multiple immune cells, including macrophages and lymphocytes [98, 99]. The interaction between PD‐1 and its two distinct corresponding ligands, programmed death ligand 1 (PD‐L1) and programmed death ligand 2 (PD‐L2) [100], play an important role in regulating the tumor environment. Overexpression of PD‐L1 is often associated with tumor metastasis and high grade gliomas [3]. One possible mechanism for the immune escape in GBM is through the interaction between PD‐1 and extracellular vesicles secreted by GBM cells [101]. Anti‐PD‐1 antibodies are one of the checkpoint inhibitors that can specifically suppress PD‐L1 activation in the immune response. Anti‐PD‐1 antibodies can promote the activation and infiltration of T cells [102] and tune up the pre‐existing immune responses [98], suggesting a potential target for prolonging the overall survival of patients.
In line with this, anti‐PD‐1 treatment has a better clinical outcome when combined with traditional therapies. For example, when the anti‐PD‐1 treatment is combined with radiotherapy, an increase in CD8+ lymphocytes [102] and macrophages, especially the portion of M1 macrophages, is clearly observed. This optimistic change in the TME improves the efficiency of the immune response in gliomas and greatly improves the overall survival [96]. The combined therapy of anti‐PD‐1 treatment and TMZ chemotherapy also has a more efficient clinical outcome than monotherapy. The median survival of mice with orthotopic gliomas was significantly improved in the combined therapy group compared to other groups, including the control group and the monotherapy groups (anti‐PD‐1 treatment or TMZ treatment), suggesting that combined therapy can promote the tumoricidal immune response to prolong survival [103].
A combination of anti‐PD‐1 and other immune checkpoint inhibitors also exhibits a novel potential therapeutic effect. CCL2, the first chemokine observed in the TME [4], is a member of the C‐C chemokines family. C‐C chemokines are associated with recruitment and polarization of TAMs, which influence tumor progression and evasion [104]. To fully exercise its immune surveillance in the CNS, CCL2 has to cooperate with its ligand, C‐C chemokine receptor type 2 (CCR2). Together, this CCL2/CCR2 axis demonstrates a significant pro‐tumor effect by encouraging tumor growth and immune suppression in the glioma TME [105]. CCX872, an antagonist of CCR2, blocks the CCL2‐CCR2 axis. CCX872 can prolong the median survival in mice with KR158 glioma by itself and demonstrates a stronger therapeutic effect in mice with 005 GSC GBM when applied with anti‐PD‐1 as a combined therapy [106].
Apart from prolonging the survival of patients with glioma as written above, immunotherapy also improves the quality of life (QoL) in patients with gliomas. Traditional treatments include surgery, chemotherapy, and radiotherapy, often with fractionated wholebrain irradiation (fWBI). However, using radiotherapy, let alone fWBI, leads not only to inhibition of tumor growth, but also to deficits in cognitive function, such as loss of memory [107]. Interestingly, combination treatment with CSF‐1R and fWBI prolongs survival and rescues the fWBI‐associated cognitive function of mice with glioma, offering potent opportunities to improve both the survival and QoL of patients with gliomas [108].
5 Conclusion and perspective
Gliomas, the most common primary brain tumors, have a poor prognosis, despite the exploration of new therapies for decades. Thus, there is a desperate demand for better therapies, which lead to a better clinical outcome in both survival and QoL, especially for patients with GBM [1, 109]. One of the most promising new therapies is immunotherapy, which demonstrates outstanding anti‐tumor effects in a series of clinical trials by arresting proangiogenic behavior and relieving the immunosuppression [1].
TAM‐associated immunotherapy has emerged as a potential therapeutic strategy. TAMs are recruited by signals produced by glioma cells or non‐malignant cells in the glioma TME and are abundant in the tumor region [110]. TAMs become polarized into either M1 or M2 phenotype and shift between the two phenotypes. The M1 and M2 macrophages exert dual influences on glioma progression by adjusting the adaptive immune responses, excessive angiogenesis, and tumor metastasis. Also, TAMs can modulate the glioma TME. M1 macrophages suppress tumor progression, while M2 macrophages encourage the neoplastic cells to completely express their malignancy. There are hundreds of macrophage phenotypes and microglia, based on their surface markers, which exert a tremendous impact on pathological and physiological changes [111 –114]. Fully understanding the interaction between these cells and their pathological and physiological impact on human bodies may also help to explain the disease to a deeper level. Overall, targeting TAMs is a promising novel therapeutic strategy for suppressing tumor progression and improving prognosis, not only as a monotherapy but also when combined with either standard therapies or immunotherapies.
Some obstacles still desperately need to be solved for TAM‐based therapies. First, resistance in TMZ‐based chemotherapy and TAM‐based immunotherapy is often observed. One of the possible mechanisms in immunotherapy is the IGF‐1R/PI3K signaling, which is believed to participate in the resistance to CSF‐1R inhibition, resulting in tumor recurrence [89]. Second, the BBB is a challenge for drugs to get access to the brain microenvironment. Under pathological contexts like GBM, BBB can be further defined as the blood–brain tumor barrier (BBTB) [115]. The BBTB has relatively narrow vasculature, which prevents peripheral drugs from entering the GBM microenvironment and exhibiting the expected pharmacological effect on GBM, especially for some new drugs like nanocarriers [116, 117].
In summary, manipulating TAMs in the TME can suppress tumor progression. Therefore, this approach may result in a durable consistent anti‐tumor response by regulating the innate and adaptive immune response, abnormal angiogenesis, and tumor metastasis. Furthermore, TAMs exhibit an excellent advantage in being more genomically stable than malignant cells, resulting in less possibility of drug resistance and treatment failure [109]. Unfortunately, although TAM‐associated immunotherapy highlights a potential opportunity for enhancing the efficacy of anti‐glioma therapy, there are no powerful and effective therapies for gliomas that are available at present. To conquer the resistance in immunotherapies and fulfill the goal of targeting TAMs as either a monotherapy or combined therapy, further exploration into the possible relationships between TAMs and neoplastic cells and the TME is of great importance.
Footnotes
Conflict of interests
The authors declared that they have no conflict of interests.