
Abstract
Endometriosis is a heritable complex disorder that is influenced by multiple genetic and environmental factors. Identification of these genetic factors will aid a better understanding of the underlying biology of the disease. In this article, we describe different methods of studying genetic variation of endometriosis, summarize results from genetic studies performed to date and provide recommendations for future studies to uncover additional factors contributing to the heritable component of endometriosis.
Keywords
Endometriosis as a complex disease
Endometriosis is a complex disorder that is caused by combination of multiple genetic and environmental factors. This means that inherited genetic variants associated with endometriosis represent only part of the risk associated with the disease. Variants confer genetic susceptibility to develop the disease but environment and lifestyle factors also play an important role, both through interaction with genetic factors and independently [1].
The heritable component of endometriosis has been illustrated by many different studies [2–4]. Higher rates of endometriosis among relatives of endometriosis cases compared with controls was shown, with risk ratio compared with general population risk for sisters estimated at 5.2 and for cousins 1.6 in a population-based study in Iceland [3]. In addition, in a hospital-based study in the UK, consisting of 230 women with surgically confirmed endometriosis in 100 families, familial aggregation of endometriosis was shown [4]. However, estimates of familial aggregation in human populations are likely to be affected – to an unknown extent – by the fact that endometriosis is only reliably diagnosed through laparoscopy. The chance of being diagnosed with endometriosis may be influenced by having a family member already diagnosed with disease and it is difficult to get an accurate population-based estimate of disease risk [5]. Stronger evidence of heritability is provided by twin studies, that have shown higher concordance in monozygotic twins compared with dizygotic twins, a finding less likely to be affected by selection biases operating on diagnostic opportunity [6,7]. The largest twin study, among 3096 Australian female twins, estimated the heritable component of endometriosis at 51% [7]. In addition to the human studies, familial aggregation of endometriosis is shown in nonhuman primates that develop endometriosis spontaneously, such as the rhesus macaque evidence, which is also less likely to be affected by diagnostic bias [8].
The underlying biological mechanisms causing endometriosis remain poorly understood to date. One fundamental approach to uncover the underlying mechanisms and causes of endometriosis is through identification and understanding of the functions of the genetic variants responsible for the heritable component of the disease.
Discovery of underlying genetic risk variants
Approaches for the investigation of genetic variants underlying endometriosis divide into hypothesis-based and hypothesis-free methods. Hypothesis-based approaches rely on prior biological understanding of the disease through selection of functionally relevant genes/regions to test for association with the disease, whereas hypo thesis-free approaches target the whole genome without preselecting any regions or genes.
Hypothesis-based candidate gene studies
Hypothesis-based candidate gene association studies have to date been the most common type of study in the investigation of genetic factors underlying complex diseases. Candidate genes studies can be either biological or positional. Biological candidate gene studies are based on prior selection of genes with a hypothesized function relevant to the disease. Variants in such genes are tested for association with disease risk. This type of biological candidate gene studies are inherently limited, as they assume a known underlying biological mechanism, a limitation that is particularly relevant in a disease such as endometriosis for which etiology is not well understood.
We previously published reviews of candidate genes studies of endometriosis, up to 1 April 2008 [9], and up to 1 April 2012 [10]. A systematic search of PubMed from 1 April 2012 to 1 December 2013 resulted in an additional 12 studies that have been published, testing 31 variants in 14 candidate genes (Table 1). Similar to other complex disease fields, candidate gene studies of endometriosis have been unsuccessful in providing replicable results [10]. For an identified association to be credible, the result needs to be replicated in an independent study in individuals from the same ethnic population. The reasons for failure of these biological candidate gene studies are now well understood. First, the tested biological hypothesis may not be true due to lack of understanding of the underlying biological mechanisms of the disease. Second, usually only a few selected variants are tested with incomplete coverage of the genetic variation in the candidate gene. Third, only the involvement of one or a few genes in a biological pathway is tested, missing potential other genes making up the pathway. Fourth, the cases and controls within and between these studies are not well defined or their definitions vary, which reduces the power to detect or replicate associations [11]. Last, the sample sizes are usually insufficient to detect genetic variants for complex traits.
In positional candidate gene studies, genes and variants are selected based on prior evidence of the likely genomic location of a disease risk variant from hypothesis-free whole genome linkage studies (described later in the article) combined with biological plausibility. This type of positional ‘filtering’ is a method that a priori has a greater likelihood of identifying genes harboring risk variants, although depending on the extent and knowledge of the region, the number of biologically interesting genes may still be large. Relatively few such studies have been performed in endometriosis [23–25].
Hypothesis-free studies
There are two types of hypothesis-free study designs: family-based linkage studies and genome-wide association studies (GWAS). The two designs are fundamentally different, but complementary, approaches in the identification of genetic risk variants across the whole allelic frequency spectrum. Linkage studies are aimed at identifying genetic variants that are rare in the general population, but are responsible for aggregation of disease in a family, whereas association studies are aimed at identifying common genetic variants in the general population related to disease risk.
Linkage studies are family/pedigree-based, and utilize the information of disease aggregation together with chromosomal recombination events, identifying chromosomal regions shared more commonly between multiple affected in a family than expected by the laws of Mendelian inheritance. They have been highly successful in identifying high-risk variants in monogenic diseases such as cystic fibrosis or Huntington's chorea [26,27]. In complex disease, however, due to underlying heterogeneity in (genetic) causality, linkage evidence tends to point at very large genomic regions that typically extend across 10–50 Mb, containing hundreds of genes. This is in contrast to association studies that can pinpoint a signal to 10–500 Kb depending on the local linkage disequilibrium (LD) structure.
A summary of genome-wide linkage studies of endometriosis to date is provided in Table 2.
Whole genome linkage studies
To conduct a linkage study of a complex disease such as endometriosis, many families with multiple affected women are required. The largest genome-wide linkage study for endometriosis was conducted by the IES, which included 1176 families from Australia (n = 931) and UK (n = 245) with at least two members – mainly affected sister pairs – with surgically diagnosed endometriosis (Table 2). This study identified a region of significant linkage on chromosome 10q26 and another region of suggestive linkage on chromosome 20p13 [33]. In a subsequent study by the IES, including a subset of 248 families with three or more affected members, an additional linkage region was identified on chromosome 7p13–15 (Figure 1) [34].
Candidate gene association studies of endometriosis published from April 2012 to December 2013, and summarized evidence for the genes from all candidate gene association studies to date.
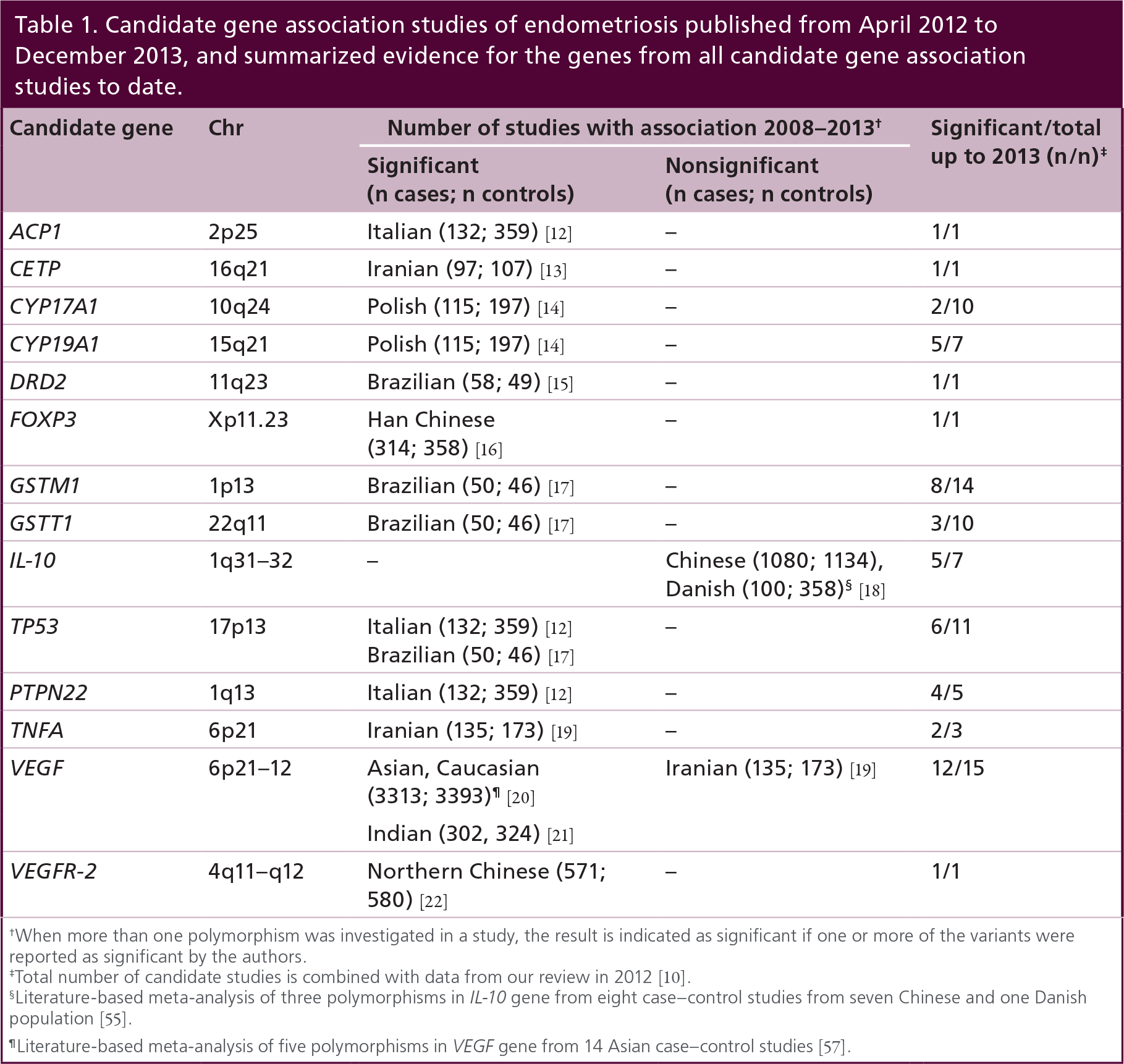
When more than one polymorphism was investigated in a study, the result is indicated as significant if one or more of the variants were reported as significant by the authors.
Total number of candidate studies is combined with data from our review in 2012 [10].
Literature-based meta-analysis of three polymorphisms in IL-10 gene from eight case-control studies from seven Chinese and one Danish population [55].
Literature-based meta-analysis of five polymorphisms in VEGF gene from 14 Asian case-control studies [57].
To follow-up these linkage regions and identify genetic variants, ‘fine mapping’ studies have been performed. The first such study extensively genotyped the linkage region on chromosome 10q26 using 11,984 single nucleotide polymorphism (SNPs) in 1144 familial cases and 1190 controls (Figure 1). The study identified three independent signals with significant evidence of association to endometriosis including rs11592737 at 96.59 Mb, rs1253130 at 105.63 Mb and rs2250804 at 124.25 Mb [28]. However, only rs11592737 was robustly replicated (p = 0.04) in an independent cohort of 2079 cases and 7069 controls, rs11592737 is located in the CYP2C19 gene, which is an important potential candidate gene for endometriosis. It is involved in drug metabolism as well as estrogen metabolism including conversion of E2 to estrone (E1) [29]. The association was unlikely to explain all of the linkage signal observed on chromosome 10q26, and further studies are needed to identify rare genetic variants in this region, through resequencing approaches. Functional studies are needed to identify the genes and determine the effects of the variants in underlying biological pathways.
A second follow-up study was performed for the chromosome 7p13–15 linkage region through sequencing of the coding regions and upstream regulatory regions of three strong candidate genes in the linkage region, namely INHBA, SFRP4 and HOXA10, with roles in endometrial development (Figure 1) [30]. The sequencing was performed in 47 cases from 15 families, aimed at identifying rare variants that are not present on genotyping arrays. The study identified 11 variants, five of which were common (minor allele frequency [MAF] >0.05) and the remaining six variants were rare. However, none of these variants either individually or collectively was significantly associated with endometriosis risk, and rare variants in the coding regions or the regulatory regions of these three genes are unlikely to be responsible for the linkage observed in these families. As only coding and regulatory regions were sequenced, it remains possible that the noncoding regions of these three genes (i.e., intronic regions) harbor rare variants associated with endometriosis, or indeed that other genes in this linkage region contain risk variants.
Genome-wide association studies
GWAS have been very successful in identifying common genetic risk variants for various complex diseases. GWAS became a reality after two major scientific and technological developments. Firstly, the International HapMap Project together with the International SNP Consortium mapped approximately 10 M common SNPs and the LD pattern between them in different ethnic populations [31], that allowed efficient genome-wide assays, through the genotyping of tagSNPs, of most of the common variation across the genome. Second, high-throughput technology generated genotyping arrays able to genotype 100,000s to millions of SNPs at ever decreasing costs. For a detailed description of the design of GWAS see Zondervan & Cardon [11]. All genetic variants identified for complex disorders and traits through GWAS are documented and regularly updated in the National Human Genome Research Institute (NHGRI) GWA Catalog [35].
To date, a total of five GWAS of endometriosis have been published in populations from European and Japanese ancestry (Table 2). The first study published was in 1432 cases (mixture of surgically diagnosed and clinically diagnosed women) and 1318 control patients from the Japanese population [32]. The discovery phase analysis included 460,945 SNPs after quality control criteria were applied. The second phase replication analysis included an independent set of 484 cases and 3974 control patients and the top 100 most significantly associated SNPs from the discovery phased were followed up. The study identified a significant association with rs10965235 located in CDKN2B-AS1 gene on chromosome 9 (p = 5.6 × 10−12; odds ratio [OR]: 1.44 [1.30–1.59]) and a suggestive association with rs16826658 near WNT4 on chromosome 1 (p = 1.7 × 10−6; OR: 1.20 [1.11–1.29]).
The second Japanese GWAS was on a smaller sample size of 696 cases (not surgically confirmed) and 825 controls including 282,828 SNPs tested after quality control [36]. This study did not discover any significant risk variants for endometriosis, which is likely to be due to a combination of the small sample size and sub optimal case definition.
The first GWAS on endometriosis among women of European ancestry was performed by an extension of the IES – the International Endogene Consortium (IEC), involving 3194 surgically confirmed endometriosis cases and 7060 controls from Australia and the UK, including 504,723 SNPs after quality control [37]. Disease severity of the cases was also categorized using rAFS classification system from retrospective surgical records, which resulted in two phenotypes: stage I and II or some ovarian disease with a few adhesions (mild disease) (n = 1686; 52.7%) and stage III and IV disease (n = 1364; 42.7%) or unknown (n = 144; 4.6%).
This study analyzed the effect of all SNPs combined showing a significantly higher ‘genetic loading’ among stage III and stage IV endometriosis cases (proportion of endometriosis variation explained by common SNPs: 0.34 [SD: 0.04]) compared with stage I and stage II cases (variation explained: 0.15 [SD: 0.15]; p = 1.1 × 10−3). Driven by this result, two GWA analyses were performed; including all endometriosis cases (n = 3194 cases), including only stage III and IV endometriosis cases (n = 1364 cases).
Summary of the published genome-wide linkage and association studies of endometriosis.

The gene name at the single nucleotide polymorphism location or the nearest gene is given for each of the associated single nucleotide polymorphisms.
GWAS: Genome-wide association studies; IEC: International Endogene Consortium.

The linkage regions and association signals identified via whole genome linkage and whole genome association studies of endometriosis.
The ‘all endometriosis’ GWAS identified two genome-wide significant associations, rs12700667 in an intergenic region, which is 88 kb upstream of a microRNA (miRNA-148a) and 290 kb upstream of NFEL2L3 on chromosome 7 (p = 2.6 × 10−7; OR: 1.22 [1.13–1.32]; rs1250248 in FN1 locus on chromosome 2 (p = 1.7 × 10−5; OR: 1.17 [1.09–1.26]). Both these associations were stronger in the stage III and IV limited analysis (p = 1.5 × 10−9; OR: 1.38 [1.24–1.53]; p = 3.2 × 10−8 OR: 1.30 [1.19–1.43], respectively). In the replication phase, the top 70 most significantly associated SNPs from the discovery phase were followed up in an independent dataset of 2392 self-reported surgically confirmed cases and 2271 controls from the Nurses' Health Study I and II in the USA. The association of rs12700667 was replicated (p = 1.2 × 10−3, OR: 1.17 [1.06–1.28]) but rs12540248 showed no evidence of replication (p = 0.57). The combined analysis of Australian, UK and USA datasets further confirmed the association of rs12700667 with endometriosis (all endometriosis p = 1.4 × 10−9; OR: 1.20 [1.13–1.27]).
This study has also investigated the significant associations reported by the Japanese GWAS of endometriosis. Their results did not show an association with rs10965235 on chromosome 9 in the European women, since this SNP is monomorphic (not variable) in individuals of European ancestry. Furthermore, no SNPs in LD with rs10965235 in the European population showed any association with endometriosis, reflecting the different genetic backgrounds of the two studied populations. rs75121902 near WNT4locus, which is in strong LD with rs10965235 in the Japanese population (R2 = 0.98), showed a strong association with stage III/IV endometriosis; a meta-analysis of all endometriosis cases (since severity of disease is not reported by the Japanese GWAS) from both GWAS datasets provided genome-wide significance of the variant (p = 4.2 × 10−8; OR: 1.19 [1.12–1.27]).
A formal genome-wide association meta-analysis of the IEC GWAS and the Japanese GWAS was subsequently performed, resulting in 4604 cases and 9393 controls from Japanese and European ancestry, and additional including an independent replication dataset of 1044 cases and 4017 controls from the BioBank Japan [38]. This meta-analysis confirmed the two associations published by the original papers and identified five additional genome-wide significant loci, namely rs13394619 in GREB1 on chromosome 2 and rs10859871 near VEZT on chromosome 12 (OR: 1.20 [1.14–1.26]; p = 5.1 × 10−13), rs4l4l819 in an intergenic region on chromosome 2 (OR: 1.15 [1.09–1.21]; p = 8.5 × 10−8), rs7739264 near ID4 on chromosome 6 (OR: 1.17 [1.11–1.23]; p = 3.6 × 10−10) and rs1537377 near CDKN2B-AS1 on chromosome 9 (OR: 1.15 [1.10–1.21]; p = 2.4 × 10−9).
A fourth GWAS analysis was published in 2013, in 2019 cases and 14,471 controls of European ancestry from the USA [39]. This study reported two additional genome-wide significant associations, namely, rs2235529 near WNT4 (in LD with rs7521902 r2 = 0.66; p = 8.65 × 10−9) and rs151976l in an intergenic region 280 kb upstream of RND3 on chromosome 2 (p = 4.70 × 10−8). However, the study did not significantly replicate the rs12700667 signal on chromosome 7 (p = 0.12).
Recently, we have conducted a meta-analysis of all genome-wide significant results from all published GWAS and replication studies in endometriosis, amalgamating the evidence for all genome-wide significantly reported loci across the studies [40]. Our study has shown robust evidence for six loci associated with endometriosis and two additional borderline associations with stage III/IV endometriosis. Furthermore, all eight loci showed stronger associations when the dataset is restricted to only more severe endometriosis cases (stage III/IV) (Table 3).
Eight genome-wide significant genetic variants robustly associated with endometriosis.

Meta-analysis p-value is from Rahmioglu et al. 2014 [40]. For rs4141819 and rs1250248 the meta-analysis p-values including only stage III/IV cases is given in parenthesis, which reach borderline significance.
Odds ratio including all endometriosis cases.
Odds ratio including only stage III/IV cases.
SNP: Single nucleotide polymorphism.
Adapted from Rahmioglu et al. (2014) [40].
Biological functions of potential genes identified for association with endometriosis.
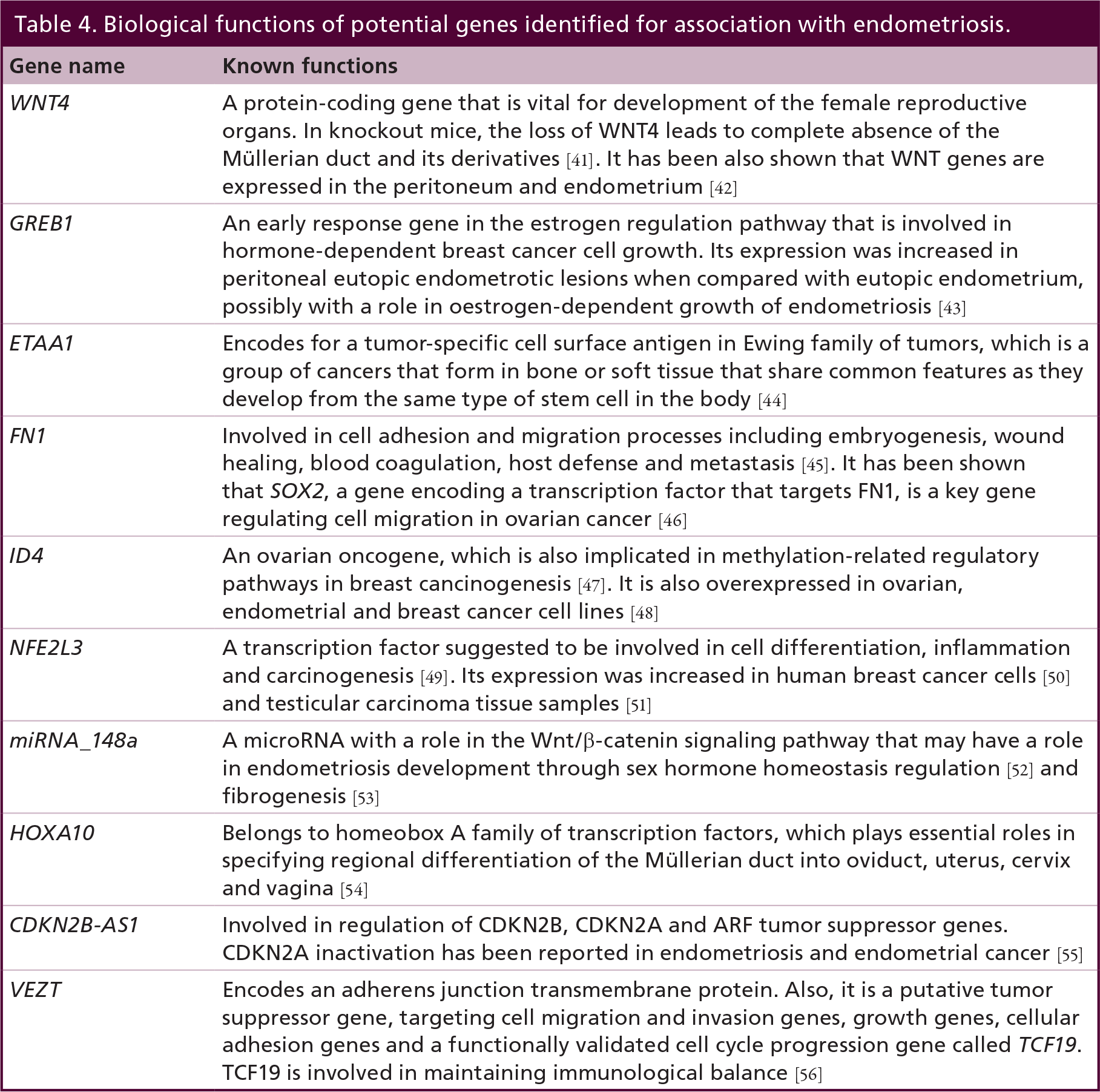
When the variants are located within genes, the biological consequences may act through these genes (e.g., for GREB1 and FN1). Alternatively, SNPs within introns or in intergenic regions may act through cis regulation of the nearby genes, or may have a regulatory action on more distal genes, including trans effects on different chromosomes. A brief summary of the functions of the genes closest to the identified endometriosis risk variants are provided in Table 4. All regions need to be followed up in functional studies to better understand their roles in endometriosis disease etiology. For an extensive description of the potential biological roles of these genes with regard to endometriosis see Rahmioglu et al. 2014 [40].
Conclusion & future perspective
The progress from genetic studies of endometriosis, and that in other complex disease fields, indicate there are a number of areas, which we can work on to improve our understanding of the genetic origins of endometriosis.
The IES results demonstrated a greater genetic loading for stage III/IV cases compared with stage I/II cases [37], along with the stronger associations observed with all the confirmed endometriosis risk variants with the more severe stages of the disease (Table 3). These results highlight the importance of collecting detailed subphenotype information on the endometriosis cases. Collection of detailed surgical data from the cases will allow assessment of genetic variants associated with different stages of the disease as well as specific subtypes of the disease such as deep infiltrating endometriosis, rectovaginal disease. Equally important is to collect the disease phenotypes in a standardized manner, which enables collaborative research and comparison of results between centers. The global WERF Endometriosis Biobanking and Phenome Harmonisation Project (EPHect) is developing freely available data collection tools to enable standardized data collection [57].
To date, the sample sizes of GWAS of endometriosis have been modest compared with other complex diseases, such as Breast cancer reaching sample size of 60,000 individuals and identifying up 68 genome-wide significant genetic risk variants [58]. Increased sample size of the GWAS meta-analysis from different populations will aid in identification of further common genetic risk variants of endometriosis [59]. This emphasizes the importance of collaboration between centers to reach larger numbers of well-defined endometriosis cases and controls.
The most commonly studied type of genetic variation is common SNPs through GWAS. However, there is a whole spectrum of rarer mutations (MAF < 0.05) both single sited and structural (e.g., copy number variations) that are likely to play a role in disease causation and explain part of the genetic heritability. These rare mutations are not captured by the genotyping arrays and are therefore missed by the GWAS. As described in the linkage studies section, there are three linkage regions identified for endometriosis, which are likely to harbor rare mutations. These regions need to be sequenced to investigate the rare mutations responsible.
It is important to note that the genetic variants identified through GWAS are unlikely to be the causal variants, but instead be in LD with the actual disease causing variants. Further sequencing or dense genotyping of these regions in large sample numbers are required to pin down the causal variants and understand their roles in downstream biological pathways. Once the genetic variants of endometriosis are identified, they need to be followed up in functional studies in relevant disease tissues such as eutopic and ectopic endometrium, to shed light on their roles in endometriosis. Functional studies include understanding the variation in different levels of the whole systems biology including, expression levels of genes (mRNA levels), protein levels and metabolite levels in different tissues and how these relate to changes in the DNA level. These will aid in understanding how the identified genetic variants cause the resulting disease phenotype.
Although mouse models of endometriosis are an important tool to better understand functional mechanisms involved in the disease [60], they are limited in that they represent an induced disease model. An important animal model in which the (genetic) origin of spontaneous disease can be studied is the rhesus macaque (Macaca, mulatta). Endometriosis develops spontaneously in rhesus macaques, and the disease is morphologically and histologically identical to that seen in humans [61]. Captive rhesus macaques are kept in colonies, which creates a smaller gene pool and greater genetic homogeneity to investigate genetic risk variants of endometriosis [61]. Furthermore, they live in controlled environments, which make them a good resource to study retrospective environmental exposures and potentially to study gene–environment interactions.
So far, the identified genetic variants of endometriosis have small effects and only explain a small proportion (3.4%) of the heritable component of the disease. Therefore, they are not suitable for use as clinical diagnostic markers. However, they provide very important starting points to investigate the underlying biological pathways contribution to endometriosis that will aid in understanding the etiology of disease. In the long term, it is hoped that they will aid in defining genetically heterogenous subtypes and allow stratification of patients for different treatments through the identification of differential pathways contributing to subtypes of the disease.
Executive summary

Endometriosis is a complex disorder with a considerable genetic component (heritability = 51 %).
Hypothesis-free genome-wide investigations of genetic variants causing endometriosis are more appropriate since the underlying mechanisms of endometriosis etiology are not well understood.
To date, eight genome-wide significant robust common genetic variants (single nucleotide polymorphisms) have been associated with endometriosis. All these associations are more strongly associated with the more moderate/severe form of the disease (rAFS stage III/IV disease).
To date, three linkage regions of endometriosis have been identified. These regions need to be followed up to identify the rare genetic variants involved that are expected to have larger effects on the disease risk.
Future studies should include more detailed and standardized disease information on the cases to allow for subphenotype analysis to identify genetic variants associated with different subtypes or groups of the disease.
Better coverage of the genomic variation in studies is crucial to investigate all of the genetic variation.
Functional studies are needed to uncover the roles of the identified genetic variants on underlying biological pathways of endometriosis.
The identified genetic variants are important in determining the underlying biology of endometriosis that will aid in developing personalized treatment options for patients.
Open access
This article is distributed under the terms of the Creative Commons Attribution License 4.0 which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited. To view a copy of the license, visit http://creativecommons.org/licenses/by/4.0/
Footnotes
GW Montgomery is supported by the NHMRC Fellowships Scheme (339446, 613667). KT Zondervan is supported by a Wellcome Trust Research Career Development Fellowship (WT085235/Z/08/Z). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.