
Abstract
Preinvasive breast carcinoma cells that proliferate and accumulate within the nonvascular, closed intraductal niche are under severe hypoxic and metabolic stress. Understanding the survival mechanisms used by these cells has revealed therapeutic strategies for killing preinvasive neoplasms. We have found that autophagy (‘self-eating’) is a major survival strategy used by preinvasive carcinoma and breast cancer stem-like cells. Based on this finding, we have opened a clinical trial that is exploring neoadjuvant oral chloroquine antiautophagy therapy for ductal carcinoma in situ. We envision that antiautophagy therapy can be administered in combination with other treatments such as those that elevate intracellular calcium, to create a state of intolerable stress for preinvasive neoplastic cells, and thereby stop breast cancer before it starts.
Keywords
Treatment of preinvasive breast lesions as a new path to breast cancer prevention
Preinvasive breast lesions proliferate within the confines of the breast duct and do not extend through the intact ductal basement membrane. One-third of all newly diagnosed breast cancer cases in the USA are preinvasive cancer [1]. Preinvasive breast neoplasms are nonobligate precursors to invasive and metastatic cancer [1–5]. Thus a subpopulation of women with preinvasive cancer will go on to develop invasive carcinoma. Compared with the general population, women harboring atypical ductal hyperplasia (ADH) and atypical lobular hyperplasia (ALH) have a 3.5- to five-fold increased risk of developing invasive cancer [1]. The increased relative risk is seven- to nine-fold for lobular carcinoma in situ (LCIS) and four- to 12-fold for ductal carcinoma in situ (DCIS) [1]. Preinvasive lesions with a higher degree of aggressive histologic features (e.g., a higher grade) have a greater risk of developing invasive cancer compared with patients with low-grade lesions [2,6–8]. Lesion size, degree of nuclear atypia and the presence of comedo necrosis (central luminal inflammation interspersed with apoptotic cells) are histopathological parameters identified as affecting the risk of recurrence within the heterogeneous spectrum of premalignant breast lesions [2,6,8]. Genetic, histopathologic and epidemiologic evidence supports that most, if not all, invasive ductal and lobular carcinomas were derived from a precursor preinvasive lesion [2,9–14]. Consequently, an intervention therapy that directly kills all preinvasive carcinoma lesions has the potential to eliminate the subset of precursor lesions that will go on to become invasive cancer.
Conventional chemoprevention embraces pharmacologic agents that block one or more early steps of carcinogenesis, initiation and genetic progression, in cell culture or animal models. Nevertheless, translation of these findings to clinical trials suffers from a 5- to 20-year waiting period to yield the reduction of breast cancer incidence in the target population. In contrast to conventional chemoprevention, we propose that a short-term therapy that kills premalignant breast lesions will prevent subsequent invasive cancer. Furthermore, such a therapy can be evaluated for safety and effectiveness as a neoadjuvant therapy for preinvasive lesions in a time period of only a few years [11,15,16].
Requirements for a therapy that targets preinvasive breast neoplasia
What are the requirements for a therapy that treats preinvasive lesions? First, the therapy must possess very low toxicity and be orally administered. These criteria are essential for targeting preinvasive lesions because no justification exists for administering a toxic intravenous therapy to a woman who may harbor occult preinvasive lesions, but is otherwise healthy. Second, the therapy must be a short course, patient administered treatment. Long-term therapies are disrupted in their effectiveness by patient noncompliance. Moreover, many therapies that show low toxicity under short-term administration exhibit significant toxicity when administered for long-term periods. If we imagine a therapy that attacks preinvasive carcinoma cells, do we want that therapy to block invasion or to directly kill the carcinoma cells? A large body of investigation has identified molecular mechanisms of invasion that constitute targets for anti-invasion therapies [17,18]. Unfortunately there are two problems with anti-invasion therapies. First, they are expected to have serious side effects, since invasion and migration is a normal part of wound healing and ongoing tissue remodeling. Second, such a therapy would only target the carcinoma cells that are in the act of invasion. Consequently, an anti-invasion therapy will have to be administered continuously for many years, since we do not know when a preinvasive lesion will unleash its invasive potential. We conclude that the ideal therapy should be a short-term therapy that directly kills the preinvasive cells.
In this article, we will demonstrate how a new understanding of preinvasive carcinoma cells survival in the nutrient deprived, hypoxic, intraductal microenvironment has led to therapeutic strategies that fulfill our criteria for the optimal short-term oral therapy that will directly kill preinvasive cells. We have found that autophagy (auto-self, phagy-eating) is a major survival strategy used by preinvasive cells within the breast duct. Based on this finding, we are currently evaluating the safety and effectiveness of chloroquine (CQ), an oral, neoadjuvant antiautophagy therapy for DCIS. We envision that antiautophagy therapy can be administered in combination with other treatments such as those which elevate intracellular calcium in the carcinoma cells, to create a state of intolerable stress for preinvasive cells, and thereby stop breast cancer before it starts.
Therapy that kills preinvasive breast lesions is a shortcut to the identification of a short-term intervention therapy that eradicates the precursors of breast cancer (

Treating preinvasive lesions is a means to prevent breast cancer.
Histopathology of preinvasive breast lesions: microenvironment constraints for intraductal neoplastic cells
Intraductal lesions are classified based on their morphologic appearance. Lobular lesions consist of cuboidal cells characteristic of luminal cells of normal breast acini, whereas ductal lesions are comprised of moderate–large columnar cells similar to normal breast duct cells [1]. The preinvasive stages of lobular breast cancer include ALH and LCIS. ALH and LCIS are considered as nonobligate precursors of invasive lobular carcinoma. The corresponding lesions of the ductal subtype include flat epithelial atypia, ADH and DCIS [1]. Flat epithelial atypia, ADH and DCIS are considered the nonobligate precursors of invasive ductal carcinoma (IDC). The pathological distinction between ADH and DCIS is based upon the degree of architectural atypia and the size and the extent of epithelial proliferation [1,6,8]. DCIS is further subclassified into low-, intermediate- and high-grade categories using histomorphological parameters that include cytomorphological and architectural features of the breast gland, such as tubule formation/differentiation, nuclear pleomorphism and mitotic index (proliferation rate) [1]. Necrosis can be a prominent feature of low- and high-grade DCIS, and the degree of necrosis correlates with the probability of invasive cancer [1,11,19].
All breast preinvasive lesions are a proliferation of neoplastic epithelial cells within the closed environment of the intraductal lumen or terminal duct lobular unit. The intraductal space in which pre invasive lesions continue to proliferate does not appear to have blood vessels or lymphatics, and is normally surrounded by myoepithelial cells (MECs) and an intact basement membrane [2,20]. The outside perimeter of the basement membrane interfaces with the connective tissue stroma, mesenchymal cells, immune cells, lymphatics and vasculature [1,2]. The pathologic hallmarks heralding the transition from in situ to invasive carcinoma are the extraductal extension of the neoplastic cells and an alteration in the cellular microecology of the lesion. By definition, preinvasive neoplasms have not yet crossed the basement membrane boundary to invade beyond their intraductal origin and then metastasize [1,21]. Microinvasion is recognized as a group of neoplastic cells that have traversed the duct basement membrane and have come into direct contact with the stroma where they can invade blood vessels, nerves and lymphatics. In addition to a disruption of the basement membrane, the cellular histomorphologic feature that distinguishes DCIS from IDC is the disappearance of the organized myoepithelial layer (
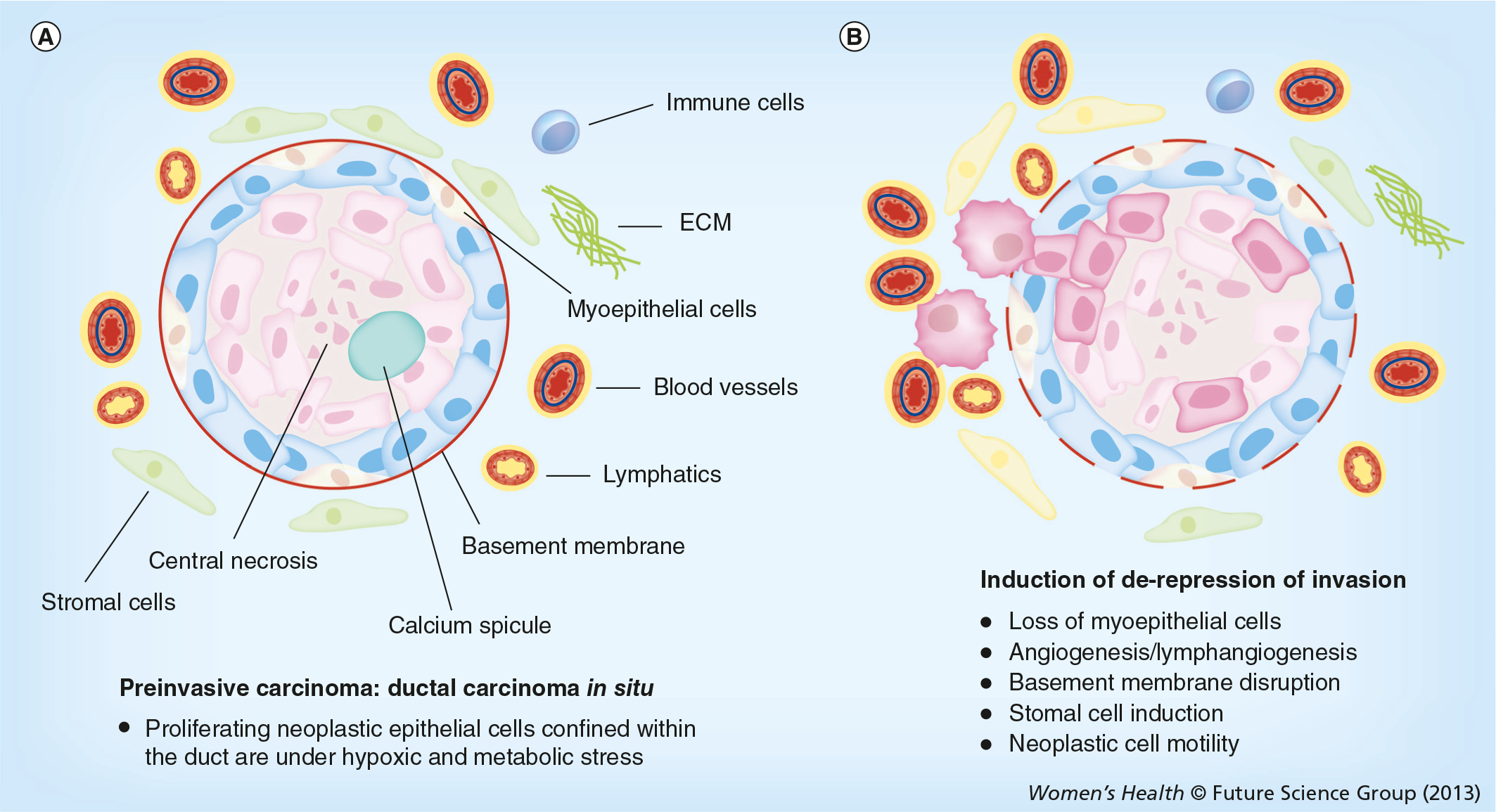
Intraductal neoplastic cells proliferate in a high-stress microenvironment
Preinvasive neoplastic cells that proliferate and accumulate within the nonvascular intraductal space are under severe hypoxic and metabolic stress. Preinvasive cells must adapt to hypoxic stress within the duct in order to survive and proliferate. The vascular density of tissues is homeostatically regulated in all tissues and all metazoan species to restrict the maximum distance between tissue cells and the nearest blood vessel. The radii of low- and high-grade DCIS lesions can be as great as 200–500 μm (15–60 cell diameters) [2]. This greatly exceeds the homeostatic limitations of blood vessel minimum density, which has been measured to fall in the range of 25–50 μm [24,25]. Multicellular spheroids grown in culture provide a model for the oxygen diffusion limitations of the nonvascularized DCIS cell colony packed within the duct bounded by the basement membrane [11,12]. Spheriods grown in culture exhibit central necrosis when the radius of the spheroid exceeds a minimum distance required for oxygen to diffuse in from the surface of the spheroid. The width of the viable rim of multicellular tumor spheroids grown in spinner culture was reported to be 150 μm over a wide range of spheroid diameters from 400 to 1000 μm [26,27]. This distance is in the range of the average distance of vessels from the nearest area of necrosis studied in solid tumors [28,29]. Therefore, typical intermediate- or high-grade preinvasive lesions expand to a diameter greatly exceeding the limitations of oxygen diffusion. High-grade preinvasive lesions may contain a central zone of necrosis with a surrounding rim of viable cells, 5–25 cell layers thick. The viable intraductal rim can exceed the minimum distance for adequate oxygenation, demonstrating that preinvasive cells have adapted to survive and proliferate under hypoxic conditions. However, adaptation to stress within the duct may contribute to the carcinogenic process [30–32].
Survival and adaptation of premalignant cells within the stressful, hypoxic and nutrient deprived intraductal microenvironment may promote genetic instability and the selection of neoplastic cells with invasive potential. Metabolic acidosis [33,34] and hypoxic stress within the tumor microenvironment induces mutagenesis and genetic instability [19,30–32]. Adaptation to survival under stress within the intraductal microenvironment can override normal cellular stress responses, leading to continued growth of genetically damaged cells.
In hypoxic conditions, DNA binding transcription factors form complexes with hypoxia inducible factors (HIFs) directing most of the adaptation. HIFs mediate the adaptive response to maintain oxygen homeostasis [28,32]. Under normal oxygen levels prolyl hydroxylases (PHD1–3) use oxygen as a substrate to modify proline residues on the oxygen dependent subunit HIF-1α. Hydroxylated HIF-α is recognized by the von Hippel Lindau tumor suppressor and thereby, targeted for proteasomal degradation [35]. Consequently, normal oxygen levels are associated with degradation of HIF-α, and conversely, HIF-α levels are increased by hypoxia. HIF-mediated adaptation to hypoxia can inhibit p53-mediated death of DNA damaged cells [32]. Hypoxic stress, independent of HIF, is associated with decreased rate of DNA damage repair, and the upregulation of invasion and metastatic potential [36–38]. Consequently preinvasive cells that adapt to living in a state of hypoxia can proliferate in the presence of genetic mutations that drive tumor progression and induce or de-repress invasion [38,39]. This stressful intraductal DCIS microenvironment is a ‘training ground’ for malignant cells (
Living in the harsh intraductal microenvironment requires cells to perform two major functions [11]: suppress pathways designed to normally eliminate damaged cells and find other ways to harness energy. Genotoxic, metabolic, hypoxic and oxidative stress engage stress-response programs in normal cells. If DNA damage is significant, then general classes of pathways are activated to eliminate propagation of damaged cells by suppressing the replication rate (senescence) or increasing the death (apoptosis) rate of damaged cells. Premalignant and malignant cells have been proposed to downregulate suppressor pathways or upregulate prosurvival pathways [6,34]. Nevertheless, even if a cell can resist programmed cell death or senescence it will not survive in a hypoxic nutrient deprived environment unless it can find alternative sources of energy for cellular functions. Preinvasive cells can exhibit a high rate of proliferation as indicated by Ki-67 staining, thus, they are avoiding senescence. Alternative pathways for obtaining energy include autophagy, anaerobic respiration or increasing the efficiency of aerobic respiration [6,34,40]. Understanding how proliferating preinvasive cells circumvent stress-induced death or cell cycle arrest, and find alternative sources of energy provides a new functional approach to create intervention strategies for premalignant breast lesions.
Preinvasive cells use autophagy to find new sources of energy under stress
Understanding cell processes, which promote survival of malignant progenitor cell in DCIS, provides strategies for killing DCIS cells, by removing their ability to survive in the stressful intraductal space. We have found that autophagy is a major mechanism used by DCIS cells to survive in the high-stress environment of the intraductal space [11,12]. We examined the hypothesis that fresh human DCIS lesions contain pre-existing carcinoma precursor cells with breast cancer stem-like cell properties, and possess invasive properties [12]. Our model system for ex vivo organoid culture of fresh human DCIS lesions, induced the emergence of neoplastic epithelial cells exhibiting the following characteristics: spontaneous generation of hundreds of spheroids (mammospheres) and duct-like 3D structures in culture within 2–4 weeks, from both low- and high-grade lesions; tumorigenicity in nonobese diabetic severe combined immunodeficient mice; cytogenetically abnormal (copy number loss or gain in chromosomes including 1, 5, 6, 8, 13 and 17) compared with the normal karyotype of the non-neoplastic cells in the source patient's breast tissue; in vitro migration and invasion of autologous breast stroma; and upregulation of signal pathways linked to, and components of, cellular autophagy and prosurvival [12].
Multiple markers of autophagy were present in the patient's original DCIS lesion and the mouse xenograft [12]. Treatment with a lysosomotropic inhibitor (CQ phosphate) of autophagy completely suppressed the generation of DCIS spheroids/3D structures, suppressed ex vivo invasion of autologous stroma, induced apoptosis, suppressed autophagy-associated proteins including Atg5, AKT/PI3 kinase and mTOR, eliminated cytogenetically abnormal cells from the organ culture and completely prevented tumor xenograft growth. Therefore, these malignant precursor cells must utilize cellular autophagy for survival [12]. Recently it has been confirmed that cultured lines of breast cancer stem cells require autophagy for survival [41]. Thus, breast cancer stem-like cells first arise within preinvasive lesions prior to the overt manifestation of invasion. Since these cells survive by the use of autophagy, this pathway constitutes an exciting therapeutic target.
What is autophagy? Autophagy is known to be a main determinant of cell fate in response to metabolic stress [40,42–45]. Autophagy (literally ‘self-eating’) denotes any cellular pathway involving the delivery of cytoplasmic material to the lysosome for enzymatic digestion. There are at least three types of autophagy: chaperone-mediated autophagy, microautophagy and macroautophagy. Chaperone-mediated autophagy is a mechanism that allows the direct lysosomal import of proteins which contain a particular pentapeptide motif [46]. By contrast, both micro- and macro-autophagy involve dynamic membrane rearrangements that snare or engulf the cytoplasmic target for loading into the lysosome. In microautophagy, cytoplasmic material is directly engulfed at the surface of the lysosome by septation, protrusion or invagination of the limiting membrane.
Macroautophagy involves the sequestration of cytoplasmic contents into a separate double-membrane cytosolic vesicle referred to as an autophagosome (

Autophagy in essence is a controlled process of self-cannibalization [47,48]. The output of the lysosomal digestion are inputs to cellular metabolism, generating energy to build new proteins and membranes. During cell starvation, autophagy provides an internal source of nutrients for energy generation and, thus, survival [45,49]. Preinvasive neoplastic cells can multiply within the nutrient and oxygen deprived intraductal space because they exploit autophagy to survive [11,12]. Invasive carcinoma cells also exploit autophagy to survive in the face of chemotherapy or molecular-targeted therapy [11,50–53].
Autophagy is triggered through diverse sources of stress impinging on preinvasive breast cancer. The first trigger is hypoxia and nutrient stress. Proliferating ductal epithelial cells accumulating within the breast duct do not have access to the vasculature outside the duct. For this reason, high-grade DCIS is associated with central necrosis, and the accumulation of lipofuschin and calcium. The activation of autophagy may divert the hypoxic DCIS cells away from apoptosis. Autophagy may also be integral to the removal of dead and dying intraductal DCIS cells, since the intraductal space is nonvascular and dead cells are not removed by immune phagocytes that are excluded from the special closed duct niche [54].
The second trigger of autophagy is anoikis [55], the activation of apoptotic cell death for cells that have been separated from their normal adhesion substratum. Autophagy has been shown to be a key regulator of survival for cells deprived of an anchoring substratum, and may play an important role for cell survival in any anchorage independent state. Normal glandular epithelial cells require attachment to, or association with, the basement membrane extracellular matrix for continued survival. During ductal hyperplasia and dysplasia epithelial cells exist at a substantial distance away from association with the peripheral basement membrane. Moreover, autophagy may assist invading carcinoma cells as they migrate into the stroma in the absence of a basement membrane anchor. A third trigger of autophagy is matrix degradation. High-grade DCIS, microinvasion and overt carcinoma invasion is associated with interruptions, remodeling and enzymatic breakdown of the basement membrane and the stromal extracellular matrix [1,21]. Autophagy may facilitate cell movement through areas of degraded matrix by the phagocytic processing of matrix breakdown fragments. A fourth trigger for autophagy is calcium. Microcalcifications are mammographic indicators of high-grade DCIS, and calcium phosphate precipitates are potent inducers of autophagy [56].
Autophagy, a new target for eliminating preinvasive carcinoma cells
CQ is an orally administered small molecule inhibitor that blocks the autophagy pathway and fulfills the criteria set forth above for the optimal therapy that targets preinvasive cancer [57–60]. Some authors have proposed that anticancer therapies should stimulate autophagy (instead of inhibiting autophagy) so that the tumor cell will eat itself to death [61]. The current data demonstrate the opposite: autophagy is a survival mechanism co-opted by preinvasive cells and breast cancer stem cells [41,51]. There are strong preclinical and clinical data justifying the use of CQ as an anticancer agent. The safety profile of CQ has been well established for long-term prophylaxis, and acute therapy of malaria worldwide [58]. CQ has been shown to suppress N-methyl-N-nitrosurea-induced mouse breast carcinogenesis [62], enhances the effectiveness of tyrosine kinase inhibitor treatment of primary chronic myeloid leukemia stem cells [50], and has been proposed as a potential means to enhance the effectiveness of tamoxifen in vitro for tamoxifen resistant breast carcinoma cells by blocking autophagy-dependent cell survival [52,63]. CQ has been proposed as a therapy for Myc-induced lymphomagenesis because CQ induces lysosomal stress that causes a p53-dependent cell death, which does not require caspase mediated apoptosis [64]. Beyond breast cancer treatment, CQ therapy has also been shown to reduce lymphoma progression [64,65], suppress melanoma invasion [66,67] and arrest pancreatic cancer in animal models [68,69], and has been employed in clinical trials of glioblastoma [70]. Potential additional primary and secondary benefits of CQ, beyond direct killing of preinvasive cells are the suppression of carcinogen-induced transformation or progression of breast epithelium, and the enhancement of tamoxifen sensitivity in estrogen-receptor-positive breast carcinoma. Finally, CQ, in contrast to a large molecule such as a therapeutic antibody, is a small molecule that can rapidly penetrate the basement membrane of the preinvasive lesions to gain immediate access to the neoplastic cells [58,71].
PINC trial
Our ongoing clinical study (clinicaltrials.gov identifier NCT01023477 [101]), is examining the safety and effectiveness of CQ phosphate (Aralen, Sanofi-Aventis, NJ, USA) administration for a 1 month period to patients with low-, intermediate- or high-grade DCIS (
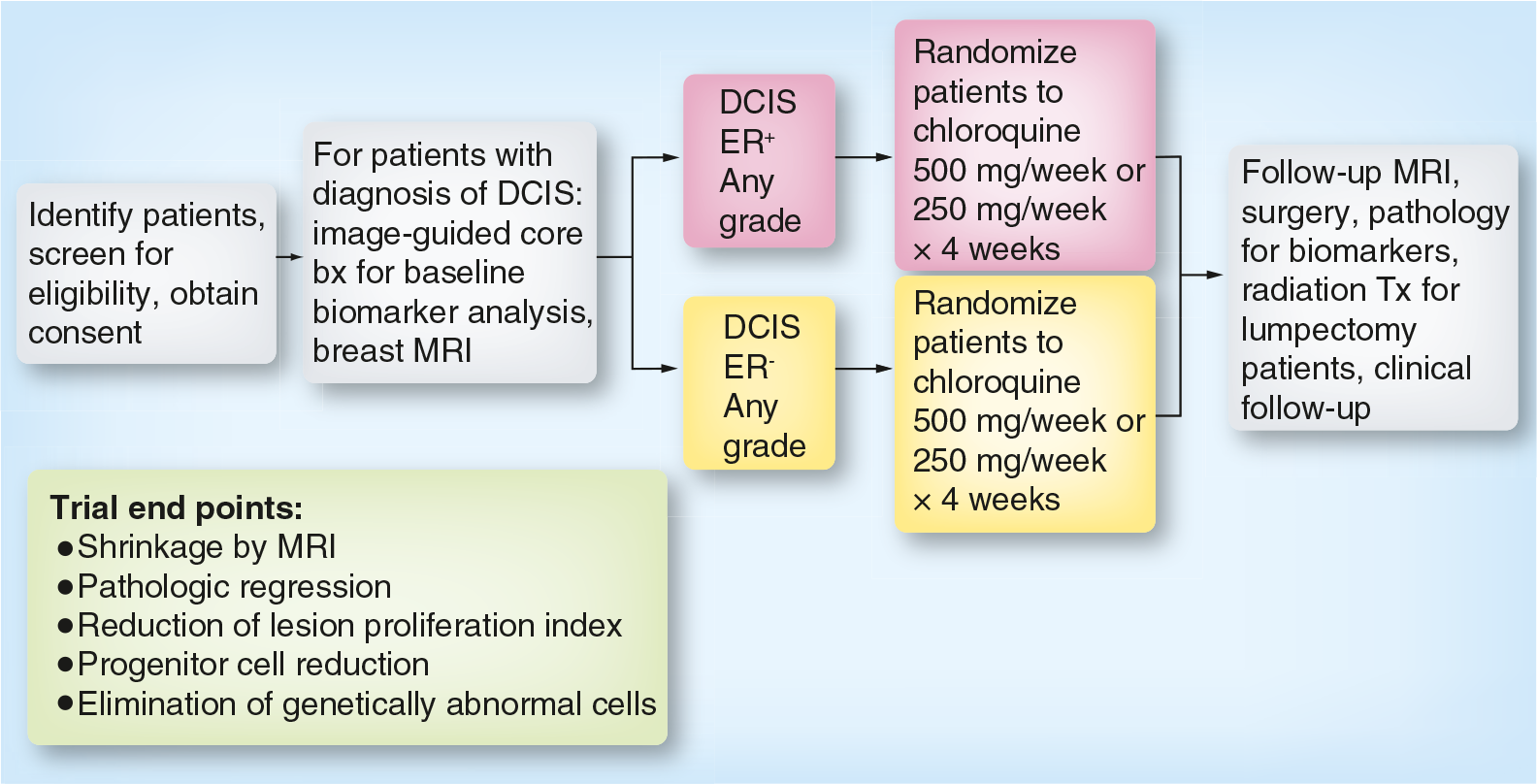
Patients with DCIS who are estrogen-receptor negative (expected to be approximately one-half of the high-grade DCIS cases) or who are estrogen-receptor positive, are both eligible for our neoadjuvant trial. Patients, regardless of histologic grade, are randomized to receive either 500 or 250 mg CQ/week for 4 weeks. MRI studies are performed on each patient at enrollment and just before surgical therapy 1 month after treatment. At the end of the 1 month treatment period, all patients receive standard-of-care surgical therapy: mastectomy or lumpectomy depending on the size and confluence of the primary DCIS lesion. Outcome measures, post- versus pretherapy, are: reduction in DCIS lesion volume by MRI; pathologic regression; the reduction or elimination of genetically abnormal tumorigenic DCIS stem-like cells; and the suppression of cellular proliferation, induction of apoptosis or disruption of autophagy, as measured by changes in proteomic markers in the post- versus pre-treatment specimen.
The design of the PINC trial has several positive features. The trial takes advantage of the standard-of-care waiting period between the initial pathologic diagnosis of DCIS and the subsequent surgical therapy. During this waiting period, CQ is administered orally. A further positive aspect is that the patient is used as her own control, because the effectiveness of the therapy will be based on a comparison of the pre- versus post-treatment specimen. In the neoadjuvant trial, we have the opportunity to evaluate the DCIS lesions radiologically before therapy, at the time of diagnostic biopsy, and then again, after therapy, prior to surgical standard-of-care excision of the neoplasm (
A final important aspect of this trial is that it can be modified to test other potential therapies, alone or in combination with CQ. Thus, the PINC trial can be a shortcut for the identification of monotherapies or combination therapies that can be documented to kill or suppress DCIS lesions following 1 month of oral therapy.
We imagine a future in which a limited course of low-toxicity therapy is administered to suppress or eradicate premalignant preinvasive breast lesions, in high-risk patients, even if they are undetectable by standard imaging. Eventually this type of therapy could be extended to the general population. While CQ is a therapeutic strategy being currently evaluated, additional antiautophagy therapies may become available in the future. Moreover, as we learn more about the survival strategies used by breast carcinoma cells, we can envision new therapies that can be used alone, or in combination with antiautophagy treatments. An exciting opportunity for combination therapy exists based on recent findings made by one of the coauthors (John Wysolmerski) concerning the importance of calcium export in breast epithelial function and breast cancer cell survival. Calcium metabolism may have a special relevance to preinvasive breast cancer progression. Pathologists and surgeons have long questioned whether intraductal calcifications, the common radiologic diagnostic feature, are a cause or consequence of breast cancer [2].
Intraductal microcalcifications record long-standing hypoxic stress
A total of 90% of DCIS mammographic diagnoses are based on the presence of microcalcifications [72,73]. While microcalcifications of all types are associated with a broad spectrum of breast lesions, and have a 30–40% overall specificity for malignancy [2], the radiologic location, shape, size and density of the microcalcification can be highly specific to DCIS [74]. Fine linear, occasionally branching microcalcifications, and pleomorphic small (<0.5 mm) calcifications, are typically found within the ductal tree, and are associated with necrotic areas of high-grade and intermediate-grade DCIS. By contrast, microcalcifications restricted to the lobules, and not the ductal tree, are almost always associated with benign disease such as microcystic adenosis. The chemical composition of most DCIS associated microcalcifications is a subtype of calcium phosphate, hydroxyapatite, which is easily detectable by conventional light microscopy. Upon histopathologic examination, microcalcifications are hematoxyphilic (blue) deposits present within the necrotic center of the DCIS lesion duct, and are often surrounded by a viable rim of DCIS neoplastic cells. The individual calcifications often appear concentrically layered, giving the impression that calcium deposition is accreting over time [75]. Suspicious microcalcifications have been associated with the later stages of fat necrosis [2], and this further supports the concept that calcium phosphate deposition follows the accumulation of necrotic cellular material. As such, microcalcifications provide an important clue about the age of intermediate- and high-grade DCIS lesions. Since hypoxia-induced necrosis precedes calcium deposition, and calcium deposition occurs over time, we can conclude that most intermediate- and high-grade DCIS lesions are subjected to hypoxic stress for a long period of time prior to diagnosis. Thus, microcalcifications can be a signature of ongoing hypoxic stress and conditions favoring genetic instability [76]. While it is unclear if microcalcifications are related to the pathogenesis of DCIS, they may contribute to the persistence of the DCIS lesions. Insoluble calcium induces autophagy, and may contribute to the local oxidative or metabolic stress within the duct [56].
Proper calcium management by breast epithelium may be a critical determinant of cell survival
An important source of cellular stress in breast epithelium is the intracellular accumulation of calcium ions that are toxic to the cell [77,78]. Export of calcium by breast epithelium, through specialized calcium export channels, is an additional strategy for preinvasive (and invasive) cancer cells to survive under hypoxic and metabolic stress [79,80].
Alterations in intracellular calcium (Ca+2) serve as important signals that modulate many cellular processes [77,78]. In order to regulate Ca+2, each cell has a Ca2+ signaling ‘toolkit’ consisting of various ion pumps, transporters and binding proteins that interact to coordinate cell- and stimulus-specific changes in Ca+2 [77,80]. One of the tools in this kit is the PMCA family, which transport Ca2+ out of the cell in response to ATP hydrolysis [81,82]. There are four PMCA genes, of which PMCA1 and PMCA4 are expressed ubiquitously. MECs transport large amounts of Ca2+ against a gradient from the systemic circulation into milk, and the cells must manage the large transcellular Ca2+ flux to avoid disruptions in Ca2+ signaling, Ca2+ toxicity and apoptosis [80]. A specific splice variant of the PMCA2 gene, PMCA2wb, which traffics to the apical membrane, is greatly upregulated in the breast at the onset of lactation, and the loss of PMCA2 reduces milk Ca2+ by 60–70% [80,83,84]. Ca2+ stimulates its own transport into milk, a process mediated by the calcium-sensing receptor, which activates the enzymatic activity of PMCA2. Thus, PMCA2 is responsible for transporting Ca2+ from MECs into milk at baseline and in response to activation of the calcium-sensing receptor. PMCA2 regulates apoptosis. During pregnancy, MECs proliferate rapidly to supply the large numbers of secretory alveoli necessary for milk production. At the end of lactation, most new MECs die in a two-step process of coordinated apoptosis [85]. Distension of alveoli by retained milk after weaning alters the shape of the epithelial cells, which, in turn, causes rapid downregulation of PMCA2 levels [79]. As a result, there is a decrease in Ca2+ export, a sustained increase in Ca+2, the activation of calpain and the onset of apoptosis. Apoptosis results from intracellular Ca2+ toxicity caused by the reduced ability of PMCA2-null cells to transport Ca2+ out of the cell when Ca2+ uptake is increased in preparation for milk production. Therefore, in addition to its importance for directional Ca2+ transport, PMCA2 also protects against cell death in response to the large influx of Ca2+ into MECs during lactation.
Calcium regulation in breast epithelium: a new target for therapies that kill preinvasive carcinoma
Intracellular Ca2+ transients regulate cellular processes important for cancer, including proliferation, adhesion, migration and apoptosis [86,87]. Ca2+ entry across the plasma membrane activates proliferation. This results both from direct effects of Ca2+ and calmodulin on cell-cycle regulators and from the activation and amplification of the Ras/Raf/MAPK pathway [88–90]. While calcium influx is tied to cell proliferation, there is a tight upper limit on the intracellular calcium concentration. Excess accumulation of Ca2+ by cancer cells induces cell death. Thus, cancer cells must reprogram intracellular Ca2+ dynamics to allow for the proliferative effects of Ca2+ entry, while preventing apoptosis from calcium overload [86,87]. Calcium export through PMCA channels, therefore, can be a major mechanism to prevent apoptosis when preinvasive cells are proliferating in a high-stress microenvironment.
1α,25-dihydroxyvitamin-D3 is the activated form of vitamin D3 and a major regulator of calcium homeostasis. We hypothesize that it can be used as a therapeutic strategy to overload preinvasive cells with Ca2+ and overwhelm the PMCA2 efflux survival mechanism, thereby promoting apoptosis. During lactation, and in cancer cells, vitamin D increases the uptake of calcium into cells through a combination of voltage-dependent and -independent channels [91–93]. However, a significant distinction is that in normal cells, vitamin D induces a transient rise in intracellular calcium levels, while in breast cancer cell lines, it induces a sustained increase in intracellular calcium and induces cell-cycle arrest in malignant breast cells, but not normal mammary epithelium in culture [93,94]. Therefore, there is strong rationale for a future strategy to kill DCIS cells by stimulating intracellular calcium uptake with vitamin D treatment while also inhibiting autophagy with CQ and/or inhibiting PMCA2 function. These combined maneuvers should induce intracellular calcium crisis specifically in DCIS lesions and convert autophagic-related and calcium export survival pathways into apoptotic death pathways [95,96]. If our hypothesis is correct, these experiments will provide the rationale for a neoadjuvant clinical trial of combination high dose vitamin D and CQ for DCIS.
Conclusion
Important sources of cellular stress in breast epithelium are nutrient deprivation, hypoxia and the intracellular accumulation of calcium ions. Our analysis of the mechanisms used by DCIS cells to survive in the hypoxic, nutrient deprived intraductal niche has revealed autophagy as a therapeutic target. Autophagy, ‘self-eating’, is a normal, cyclic cellular process designed for producing energy during cellular stress. As a survival strategy, breast epithelium upregulate autophagy and increase export of calcium. The intraductal preneoplastic cellular niche appears to be preferentially sensitive to short term, antiautophagy-based prevention therapy, which targets survival pathways used by preinvasive carcinomas.
The PINC trial, a Phase I/II clinical trial, is currently enrolling patients to assess the efficacy of an autophagy inhibitor, CQ phosphate, in reducing the viability of DCIS cells and their ability to become invasive. Our unique trial design offers a means to functionally screen investigational agents that kill premalignant breast lesions.
We imagine a future in which a limited course of low toxicity therapy is administered to suppress or eradicate preinvasive breast lesions, in high-risk patients, even if they are undetectable by standard imaging. Additional anti-autophagy therapies may become available in the future, or CQ may be combined with other therapies, providing multidimensional cell inhibition.
Future perspective
We speculate that, in the next 10 years, a new type of oral breast cancer prevention therapy that kills or suppresses preinvasive lesions will be available for use in high-risk patients, and eventually in all women in the postreproductive years. It will be a low-toxicity therapy administered for a short time (1 month), and this course of therapy may be repeated every 2–5 years. We are currently studying the first generation of this therapy, CQ, which disrupts autophagy, as a neoadjuvant therapy for DCIS and ADH (NCT01023477 [101]). The outcome of this trial should be known within the next 2 years. If we are successful in showing that CQ will reduce the radiologic size of the DCIS lesions, or reduce proliferation, and perhaps increase apoptosis, of the intraductal neoplastic cells, after the 30-day treatment course, then this will set the stage for wider confirmatory studies by others. The molecular basis of this new class of prevention will take advantage of growing scientific knowledge regarding the mechanisms used by preinvasive carcinoma cells to survive and proliferate in the high-stress microenvironment of the hypoxic and nutrient-deprived breast intraductal niche. The biologic mechanism of this new form of therapy will be different than what has been envisioned for chemoprevention agents in the past. Instead of blocking early stages of the carcinogenic process by long-term therapy, the new short-term therapy will selectively kill or suppress already transformed genetically abnormal cells that have arisen within the breast duct as precursor lesions of invasive breast cancer.
Executive summary
Preinvasive breast neoplasms are nonobligate precursors to invasive and metastatic cancer.
Preinvasive breast carcinoma cells that proliferate and accumulate within the nonvascular closed intraductal niche are under severe hypoxic and metabolic stress. Understanding the survival mechanisms used by these cells has revealed therapeutic strategies for killing preinvasive neoplasms before they can invade out of the duct.
Short-term treatment of preinvasive breast lesions is a new path to breast cancer prevention.
The ideal treatment agent is an orally administered small molecule with very low toxicity.
Autophagy is a major survival strategy used by preinvasive cells and breast cancer stem-like cells. Our findings indicate that breast cancer stem-like cells that exist within invasive carcinoma first arise within preinvasive lesions prior to the overt manifestation of invasion. Since these cells survive by the use of autophagy, this pathway constitutes an exciting therapeutic target.
Chloroquine (CQ) is an orally administered small molecule inhibitor that blocks the autophagy pathway. The safety profile of CQ has been well established for long-term prophylaxis and acute therapy of malaria worldwide.
All breast preinvasive lesions are a proliferation of neoplastic epithelial cells within the closed environment of the intraductal lumen or terminal lobular unit.
Ductal carcinoma in situ (DCIS) can be distinguished from invasive ductal carcinoma by the disappearance of the organized myoepithelial layer.
Preinvasive neoplastic cells that proliferate and accumulate within the nonvascular intraductal space are under severe hypoxic and metabolic stress.
Adaption to survival under stress within the intraductal microenvironment can override normal cellular stress responses, leading to continued growth of genetically altered cells.
Living in the harsh intraductal microenvironment requires cells to perform two major functions: suppress pathways designed to normally eliminate damaged cells, and find alternate cellular methods of harnessing energy.
Autophagy (literally ‘self-eating’) is a controlled process of self-cannibalization. Cells capture and consume their own cytoplasm and organelles. The output of the lysosomal digestion is energy, providing ATP for cellular metabolism.
During cell starvation, autophagy provides an internal source of nutrients for energy generation and, thus, survival. Preinvasive neoplastic cells can multiply within the nutrient- and oxygen- deprived intraductal space because they exploit autophagy to survive.
Invasive carcinoma cells also exploit autophagy to survive in the face of cytotoxic stress caused by chemotherapy or molecular targeted therapy.
CQ can rapidly penetrate the basement membrane of the preinvasive lesions.
CQ provides a potential therapy for estrogen-receptor-negative breast DCIS lesions.
Based on our finding that autophagy is a target for preinvasive cancer, we are currently evaluating the safety and effectiveness of CQ, an antiautophagy oral neoadjuvant therapy for DCIS in the PINC trial.
As we learn more about the survival strategies used by breast carcinoma cells, we can envision future therapies that can be used alone, or in combination with, antiautophagy treatments.
Therapy that kills preinvasive breast lesions is a short-cut to the identification of a short-term intervention therapy that eradicates the precursors of breast cancer. A treatment that kills preinvasive breast lesions follows the same rationale used for the prevention of colorectal cancer by colon polypectomy, and cervical cancer by ablating cervical dysplasia.
Intraductal calcium spicules are radiologic hallmarks of preinvasive breast cancer. An exciting opportunity for combination therapy exists based on recent findings concerning the importance of calcium export in breast epithelial function and breast cancer cell survival.
We propose a future strategy to kill preinvasive cells by stimulating intracellular calcium uptake with vitamin D treatment, while also inhibiting autophagy with CQ and/or inhibiting breast epithelial calcium export function. These combined maneuvers should induce intracellular calcium crisis specifically in DCIS lesions and convert autophagic and calcium export survival pathways into apoptotic death pathways.
We imagine a future in which a limited course of low-toxicity therapy is administered to suppress or eradicate premalignant preinvasive breast lesions, in high-risk patients, even if they are undetectable by standard imaging.
In this article, we demonstrate how autophagy appears to be an ideal target because preinvasive carcinoma cells may have become addicted to this form of self-cannibalization to generate energy needed for proliferation under stress. We further envision that other therapies can be combined with antiautophagy therapy, such as modulators of calcium efflux, so as to enhance the therapy effectiveness, and reduce the number of repeated treatments over a women's lifetime.
Footnotes
The work described herein was supported by grants to LA Liotta from the Department of Defense Breast Cancer Research Program (US Army Medical Research Acquisition Activity) and the Susan G Komen Foundation. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.