
Abstract
Drug-delivery carriers represent an important step in the development of targeted therapy. Encapsulation of drug into liposomes represents such a carrier, and helps to minimize side effects of conventional doxorubicin by improving the tumor-specific biodistribution profile. We review the development of the two liposomal doxorubicin formulations, pegylated liposomal doxorubicin and liposomal-encapsulated doxorubicin citrate from reconstitution and comparative pharmacokinetics to pivotal Phase III trials, with special emphasis in breast cancer. The relative differences in the toxicity profile can be attributed to their differences in the liposomal formulations. Areas of special interest include the reduction in cardiac toxicities and the improved efficacy, such as in the treatment of ovarian cancer. These improvements have also increased the potential of these liposomal formulations of doxorubicin for combination and sequencing with other biological and cytotoxic agents for clinical benefit.
Doxorubicin has played an important role in the treatment of breast cancer for many decades, but its clinical utility is limited by its cardiotoxicity. A retrospective analysis of three Phase III trials of patients treated with doxorubicin in combination with other cytotoxic agents or radiation therapy suggested that doxorubicin-associated cardiac events may occur more frequently and at lower cumulative doses than previously reported [1].
Drug-delivery carriers for chemotherapeutic agents represent an important step in the development of targeted therapy with enhancement of both selectivity and antitumor efficacy. Encapsulation of drug into liposomes represents one of these attempts, which may minimize the side effects and enhance the antitumor efficacy by altering its pharmacokinetics and biodistribution profile. Amphipathic molecules such as anthracyclines (doxorubicin and daunorubicin) are well suited for such liposomal encapsulation. The seminal review by Drummond and colleagues outlines certain advantages of liposome-encapsulated anthracyclines for targeted therapy. This formulation reduces the volume of distribution (Vd) and clearance and prolongs half-life (t1/2), resulting in prolonged release of the drug within the tumor environment. There is increased stability owing to limited conversion to aglycones or secondary alcohol metabolites. The drug selectively accumulates in tumors characterized by discontinuous ‘leaky’ microvasculature, or in organs containing macrophages, such as the reticuloendothelial (RE) system (liver and spleen), while sparing healthy tissues such as the heart which have a normal endothelial barrier. It also partially circumvents P-glycoprotein (Pgp)-mediated tumor resistance [2].
There are two liposomal doxorubicin formulations currently in clinical use for the treatment of breast cancer: liposomal-encapsulated doxorubicin citrate (Myocet®/D-99) (LEDC), and pegylated (STEALTH®) liposomal doxorubicin (Doxil®/Caelyx®) (PLD).
Formulation of PLD & LDEC
Doxorubicin hydrochloride (C27H29N1O11-HCl, molecular weight 579.99), the active ingredient in liposomal doxorubicin, is enclosed in an aqueous space surrounded by a lipid bilayer specific to the type of liposome. There are three lipid components in PLD:
Hydrogenated soy phosphatidylcholine (HSPC)
Cholesterol
Distearoyl-phosphatidylethanolamine (DSPE) conjugated to polyethylene glycol (PEG) HSPC/Chol/PEG-DSPE, in a ratio of 56:39:5. The lipid constituents of LEDC are egg phosphatidylcholine (eggPC) and cholesterol (eggPC/chol) in a ratio of 55:45 [2,3].
Reconstitution of the two liposomal doxorubicin formulations is totally different, with the preparation of LEDC being more complicated than PLD. LEDC is supplied as three vials containing, as follows:
Unilamellar liposomes composed of eggPC cholesterol (55:45 mol/mol), with a median diameter of 180 nm. The concentration is 100 mg of total lipid/ml of citric acid buffer (300 mM pH 4.0) in a total volume of 3.9 ml;
0.5 M sodium carbonate (pH 11.4);
Doxorubicin HC1 (with methylparaben) as a lyophilized powder. The doxorubicin is encapsulated in the liposomes by a procedure in six stages, which include heating of the solution to a temperature of 55–60°C [4].
In comparison, PLD is supplied as one vial containing 16 mg/ml of lipid with 2 mg of doxorubicin in 1 ml of solution, which is ready for reconstitution and clinical use [3].
Comparative pharmacokinetics of PLD & LDEC
The differences in the liposomal formulations confer very different biologic properties. LEDC is predominantly taken up from the circulation in the reticuloendothelial system, while pegylation generally protects liposomes from uptake by phagocytic cells and tissues, and thus results in extremely long intravascular circulation times. It has been hypothesized that pegylation enhances passive uptake of doxorubicin liposomes into tumors. However, this has to be balanced against the possibility that larger molecules such as polyethylene glycol on the liposomal surface may reduce the interaction of liposomes with cells [5,6].
Several studies have shown that the pharmacokinetics, biodistribution and toxicities of liposomal doxorubicin depend upon the type and size of liposomes. LEDC differs from PLD in having a larger mean diameter (160 vs 100 nm), a larger Vd (18.8–34.2 versus 4l) and a shorter plasma t1/2(16.4–52.6 h versus 45.9–99 h) [4,7,8–10]. Both slow-release LEDC and PLD have a Vd not significantly different from the total blood volume, indicating that the drug is generally confined to the systemic circulation. However, after intravenous administration, LEDC has saturable, nonlinear kinetics, whereas PLD has nonsaturable, log-linear kinetics [11,12].
Metabolism & elimination of liposomal doxorubicin
Anthracyclines are metabolized in human plasma to a variety of both active and inactive metabolites. The reduction in doxorubicin by an aldo–ketoreductase results in the formation of the most prominent metabolite, doxorubicinol in plasma, bile and urine. A two-electron reduction in doxorubicin with subsequent elimination of the sugar results in the inactive metabolite, a 7-deoxyaglycone [13].
The presence of doxorubicin metabolites in plasma and urine after the administration of the two forms of liposomal doxorubicin has been investigated. Although several of the more common metabolites (doxorubicinol and glucoronide/sulfate derivatives of 4-dimethyl,7-deoxy-aglycones) were observed in urine, they were at diminished levels (2.5%) compared with the administration of free doxorubicin (11%). Liposomal doxorubicin (>1 h–5 days) was eliminated at much slower rate than free doxorubicin (15 min–48 h) [7,14]. In two separate studies, doxorubicinol was not observed in plasma at any time after the administration of Stealth liposomal-doxorubicin [7,15].
The accumulation of liposomes in tumors is favored not only by a leaky microvasculature but also by an insufficient lymphatic drainage. The combination of these two factors is referred to as the enhanced permeability and retention (EPR) phenomenon [2]. The tumor microenvironment contributes to destabilizing the lipid carrier through the action of the slightly acidic pH of interstitial fluids, the release of lipases from dying tumor cells and the release of enzymes and oxidizing agents by tumor-infiltrating inflammatory cells [16].
Toxicity profile
The difference in pharmacokinetics is reflected in the different toxicity profiles. LEDC does not produce palmar–plantar erythrodysesthesia (PPE) and has myelosuppression as its dose-limiting toxicity, while with PLD, dose-limiting toxicitys are mucocutaneous reactions such as PPE and mucositis/stomatitis [17,18].
The current hypothesis for the development of PPE is that the small size (100 nm diameter) and long circulation time (t1/2 is approximately 48 h in humans) of PLD allows liposomes to accumulate in the skin. The basal layers of the skin are damaged with prolonged exposure to doxorubicin as the liposomes slowly release their contents. The accumulation of liposomes is thought to mimic the anatomical distribution of lesions and to be greatest in regions of skin that are subjected to pressure or irritation, such as the flexure creases of the hands, soles of the feet, or belt lines [18,19]. This hypothesis is supported by experimental and clinical data. Liposomes with long circulation times accumulate in the skin of experimental animals to a greater extent than liposomes with shorter circulation times [20]. In metastatic breast cancer, patients treated with PLD, changes in dose schedule have a significant impact on the toxicity outcome, even when the dose intensity is kept constant. Mucositis is the main dose-limiting toxicity for high doses, whereas PPE is the predominant toxicity for those treated at short dose intervals of 3 weeks [21]. Furthermore, grade II alopecia, which is common with doxorubicin, is rare with liposomal formulations [4,17,19].
Hypersensitivity reactions represent a problem with PLD, with a reported frequency between 0 and 45%. Unlike type I allergy, these reactions occur at first exposure without sensitization [17,22,23]. There is a significant association of complement activation and hypersensitivity, but other unidentified factors may also play a role [23].
Cardiac toxicity
Several studies indicate that the risk of anthracycline-induced cardiotoxicity is considerably lower with liposomal doxorubicin formulations (both pegylated and nonpegylated) in comparison with doxorubicin [21,24–26].
In a Phase III study, Harris and colleagues compared the cardiac safety of LEDC with that of doxorubicin in 224 patients with metastatic breast cancer and a cumulative lifetime adjuvant doxorubicin dose 300 mg/m2 [24]. Patients received LEDC 75 mg/m2 or doxorubicin 75 mg/m2 as 1 h infusions given every 3 weeks. Patients who received LEDC experienced significantly fewer cardiac events than those receiving doxorubicin (13 vs 29%, respectively, p = 0.0001), and significantly fewer patients treated with LEDC developed congestive heart failure (CHF) (2 vs 8% of patients receiving doxorubicin, p = 0.0001). The median cumulative doxorubicin dose at the onset of cardiotoxicity was significantly higher in patients treated with liposomal doxorubicin: 785 mg/m2 compared with 570 mg/m2 in patients receiving doxorubicin p = 0.0001; hazard ratio (HR): 3.56.
The cardiac safety of PLD was confirmed in a large-scale prospective study conducted by Wigler and colleagues. A total of 509 patients with no prior history of heart disease received 1 h infusions of either PLD, 50 mg/m2 once every 4 weeks or doxorubicin 60 mg/m2 once every 3 weeks. Cardiac safety was measured using multiple gated acquisition (MUGA) scans to assess left ventricular ejection fraction (LVEF) at baseline and throughout the study. The risk for developing a cardiac event was significantly lower in patients treated with PLD than in patients treated with doxorubicin (p < 0.001; HR: 3.16). Among all treated patients, at cumulative doses of less than 500–550 mg/m2, the risk for developing a cardiac event was 11% with PLD, compared with a 40% risk in the doxorubicin group. In the group of patients with high risk of developing cardiotoxicity, the risk of developing a cardiac event was significantly lower for patients treated with PLD than for those treated with the conventional formulation [27]. Interestingly, data from Kaposi's sarcoma have shown that a higher cumulative dose can be achieved with PLD compared with doxorubicin without major cardiac events [28]. For example, in one study, a very high cumulative dose of 1040 mg/m2 was achieved in one patient with no cardiac toxicity [29].
Clinical experience with LEDC in breast cancer
Phase I studies
In Phase I trials, LEDC was tested at different dose levels and schedules either alone or in combination with other chemotherapeutic agents (
Phase I studies for liposomal doxorubicin.

G-CSF: Granulocyte colony-stimulating factor; LEDC: Liposomal doxorubicin citrate; PLD: Pegylated liposomal doxorubicin; PPE: Palmar–plantar erythrodysesthesia.
Phase II studies
Several Phase II studies have evaluated the efficacy and safety profile, including the risk for cardiac toxicity, of LEDC in metastatic breast cancer and in the neoadjuvant setting in locally advanced primary breast cancer (LAPC) as monotherapy or in combination (Table 2). The results of these studies support the use of LEDC for these indications. The response rate (RR) ranged from 65–80% with clinical complete response (CR) ranging between 5 and 27%. The most common toxicities were mainly hematological. Although grade 3–4 neutropenia occurred in 9 to 61% of the patients, including febrile neutropenia in 2.7–28%, it was uncomplicated and rapidly reversible. Other nonhematological toxicities were mild with rare grade 3–4 mucositis, nausea, asthenia and arthralgia [33–39].
Phase II and III liposomal-encapsulated doxorubicin (Myocet®/D99) in locally advanced primary breast cancer and metastatic breast cancer.
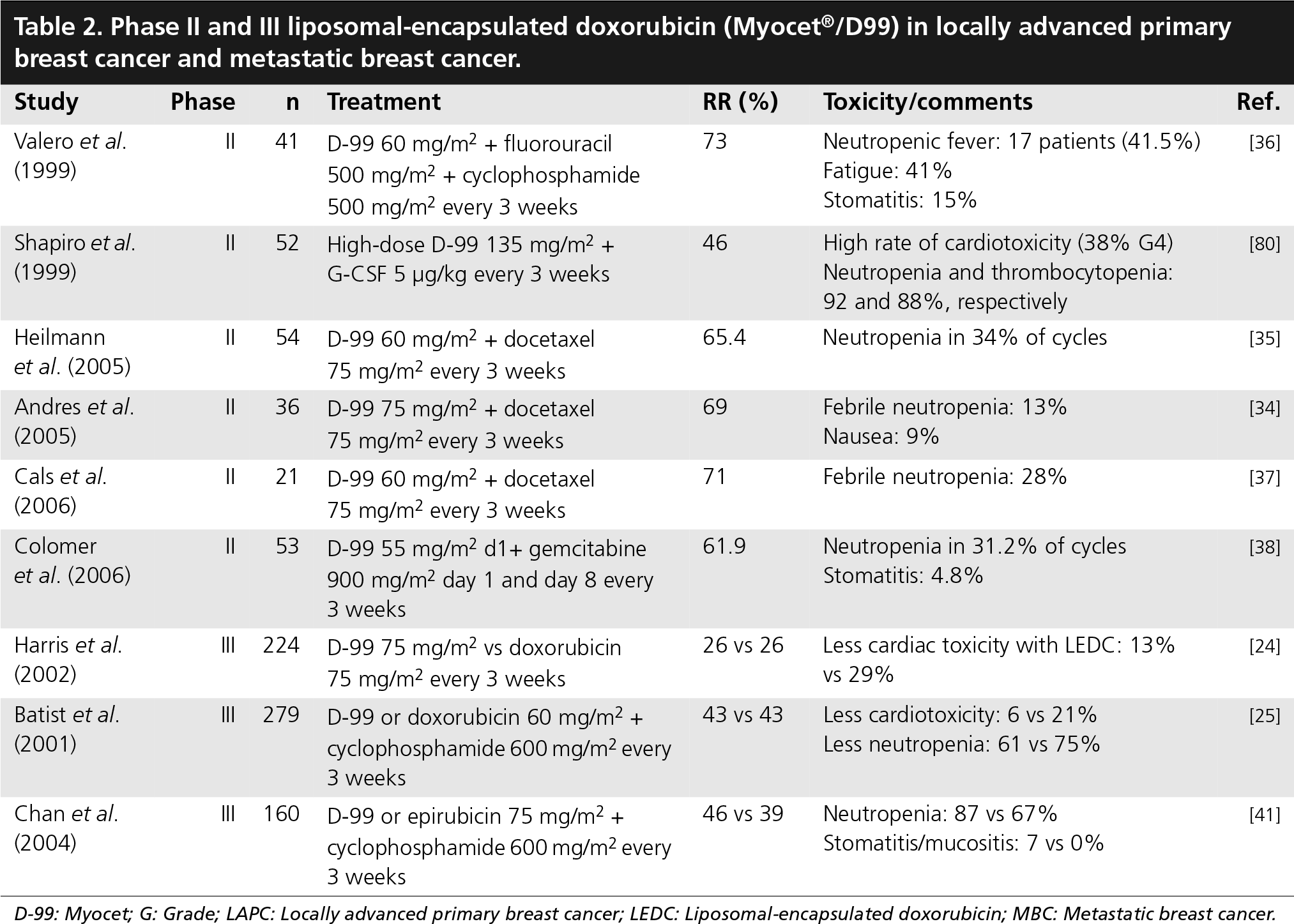
D-99: Myocet; G: Grade; LAPC: Locally advanced primary breast cancer; LEDC: Liposomal-encapsulated doxorubicin; MBC: Metastatic breast cancer.
Phase III studies
Phase III trials were conducted to compare LEDC with doxorubicin as monotherapy or in combination, with no difference in RR and a significant reduction in the risk of cardiac toxicity (Table 2) For example, in a randomized, multicenter trial to test this hypothesis, in the setting of first-line treatment of metastatic breast cancer, the RRs for both monotherapy treatment arms were the same at 26%. In the safety analysis, cardiac events that led to patient withdrawal from the study were more than twice as frequent in doxorubicin-treated patients as LEDC-treated patients (29 vs 13%, log-rank p = 0.0001). A Kaplan–Meier estimate of the probability of the first onset of a cardiac event was related to the lifetime cumulative dose of doxorubicin or LEDC, and the risk of onset of cardiotoxicity at the same cumulative dose was much higher with doxorubicin treatment than LEDC (HR: 3.56); (p = 0.0001) [24].
In the randomized, Phase III, multicenter trial in metastatic breast cancer that utilized the combination of LEDC and cyclophosphamide (MC) in comparison with the doxorubicin and cyclophosphamide (AC) combination, a CR or partial response (PR) was observed in 43% of patients in both treatment groups. Among doxorubicin-naive patients the RRs were comparable: 42% in the MC-treated group versus 45% in the AC-treated group. In the small subset of patients (10%) that had received prior doxorubicin, the objective RR was 50% for the MC group compared with 20% in the AC group. A total of 6% of the patients treated with MC developed protocol-defined cardiotoxicity, compared with 21% of those treated with AC (log-rank p = .0001). Five cases of CHF, all in the AC arm (log-rank p = 0.02), were observed after cumulative lifetime doses ranging from 360–480 mg/m2. The estimated (Kaplan–Meier) median cumulative lifetime dose of doxorubicin at the first occurrence of protocol-defined cardiac toxicity was more than 2220 mg/m2 for the MC arm versus 480 mg/m2 for the AC arm with a HR of 4.8 [25]. The improvement in cardiac toxicity in LEDC-treated patients versus doxorubicin-treated patients was also confirmed in a retrospective analysis based on pooled data from the aforementioned Phase III trials [40].
When LEDC was compared with epirubicin in a Phase III trial in combination with cyclophosphamide as first-line therapy for metastatic breast cancer, it was found that the combination that contains LEDC had a trend for higher RR (46 vs 39%, p = 0.42) and longer median survival time (18.3 vs 16 months, p = 0.504), but the data were not statistically significant. However, the median time to disease progression was significantly longer in the LEDC arm as compared with the epirubicin arm (7.7 vs 5.6 p = 0.02). However, the liposomal combination showed higher neutropenia and stomatitis/mucositis rates compared with epirubicin and cyclophosphamide. The cardiac toxicity was low in both treatment groups, as nine patients in the liposomal combination and eight patients in the epirubicin and cyclophosphamide arm developed asymptomatic LVEF reductions at comparable cumulative doses. It was concluded that LEDC appeared to be an acceptable substitution to epirubicin as first-line treatment for patients with metastatic breast cancer [41].
Other tumor sites
Apart from breast cancer, liposomal doxorubicin has been investigated in hematological malignancies such as non-Hodgkin's lymphoma (NHL). Phase II studies of LEDC in NHL generated encouraging results with proved efficacy and safety in elderly patients and patients with aggressive lymphoma, but there are no published Phase III results [42,43]. In preclinical studies, there is evidence that LEDC is preferentially taken up by the RE system, while pegylation reduces such uptake [44]. Whether this differential uptake justifies the use of LEDC in preference to PLD in NHL (and perhaps even doxorubicin in the future) remains to be investigated further.
Clinical experience with PLD in breast cancer
Phase I studies
Several Phase I clinical studies have been conducted to determine the MTD and dose-limited toxicities with PLD using different schedules and doses either as monotherapy or in combination in patients with Kaposi's sarcoma, breast cancer, ovarian cancer or other solid tumors. These studies are summarized in Table 1. A joint report was presented from two Phase I studies [17]. The results from both studies suggest that the recommended median dose intensity of PLD for Phase II trials is 15 mg/m2 per week. The MTD was established at 50 mg/mg2 every 3 weeks and at 60 mg/m2 every 4 weeks. The major adverse events were mucositis, in the form of stomatitis–pharyngitis, and skin toxicity in the form of hand–foot (H–F) syndrome (the H–F syndrome, or chemotherapy-associated PPE, is a painful, desquamating dermatitis primarily affecting hands and feet) These were the dose-limiting toxicities of PLD [17].
A Phase I study that included 44 patients with a variety of solid tumors examined the combination of PLD and weekly paclitaxel (wPTX) with an initial dose of 50 mg/m2 weekly paclitaxel and 30 mg/m2 PLD. The paclitaxel dose was escalated in increments of 10 mg/m2 and PLD in increments of 5 mg/m2 until the MTD was reached. The MTD was 30/90 and 35/80 mg/m2for PLD/weekly paclitaxel. Dose-limited toxicities included treatment delay for grade 3 neutropenia, febrile neutropenia, PPE and deep vein thrombosis [45].
A Phase I study of PLD titrated to MTD starting with 20 mg/m2 on day 1 and gemcitabine 800 mg/m2 on day 1 and 8 every 3 weeks in patients with metastatic breast cancer the MTD was 24 mg/m2 for PLD and the dose-limited toxicities were granulocytopenia and thrombocytopenia [46].
Phase II studies
The efficacy and toxicity of PLD were evaluated in Phase II studies in patients with metastatic breast cancer, in both anthracycline-naïve patients or in patients with prior anthracycline treatment, and in the neoadjuvant setting in patients with LAPC.
In single-agent studies, the RR was relatively low (~30%) and toxicities included mucositis, PPE and neutropenia [38,47–49].
One of these studies evaluated the clinical benefit of PLD in patients with metastatic breast cancer, previously treated with conventional anthracyclines. A total of 79 metastatic breast cancer patients received PLD 50 mg/m2 every 4 weeks. The overall clinical benefit rate (objective response plus stable disease 24 weeks) was 24% (16.1% in patients with documented anthracycline resistance vs 29% in nonresistant patients). It was concluded that the use of PLD is associated with an obvious clinical benefit in patients previously treated with conventional anthracyclines, and this clinical benefit is highest in patients with less exposure to prior chemotherapy regimens and taxane-naive patients, and lowest in patients with documented anthracycline resistance [47].
A retrospective analysis of two European Organisation for Research and Treatment of Cancer (EORTC) studies evaluated the impact of age, using a 70-year cut-off, on the safety and efficacy of PLD given at 60 mg/m2 every 6 weeks (treatment A) or 50 mg/m2 every 4 weeks (treatment B) to 136 metastatic breast cancer patients, of whom 65 were 70 years of age or older. No difference in terms of toxicity was observed between younger and older patients treated with the 4-week schedule, while a higher incidence of hematological toxicity, anorexia, asthenia and stomatitis was observed in older patients when the 6-week schedule was used. Antitumor activity was not affected by age. In the older cohort of patients, no dependence was found between the incidence of grade 3–4 toxicity or antitumor activity and patients’ baseline performance status, comorbidities or concomitant medications. The higher therapeutic index of Caelyx 50 mg/m2 every 4 weeks makes it, of the two dose schedules investigated, the preferred regimen in the elderly [50].
The efficacy of PLD was further enhanced by combination with other chemotherapeutic agents that have different mechanisms of action and nonoverlapping toxicities. This has been documented in several studies in combination with cyclophosphamide [51,52], vinorelbine [53,54], gemcitabine [55,56], taxanes and trastuzumab [57,58,59]. The RRs range from 33–75%, which is higher than single agent-studies. The adverse events associated with these combinations were neutropenia, PPE, stomatitis and infusion reactions, but most of these were tolerable with no toxic deaths and noticeable cardiac safety. The results of these studies are summarised in Table 3.
Phase II & III pegylated liposomal doxorubicin (Doxil®/Caelyx®) in locally advanced primary breast cancer and metastatic breast cancer.

G: Grade; LAPC: Locally advanced primary breast cancer; LEDC: Liposomal-encapsulated doxorubicin; MBC: Metastatic breast cancer; PLD: Pegylated liposomal doxorubicin; PPE: Palmar–plantar erythrodysesthesia.
Phase III studies
In a Phase III study that was designed as a noninferiority trial to assess treatment efficacy and cardiac safety of PLD as compared with doxorubicin as first-line therapy for metastatic breast cancer, the results showed that both drugs were comparable in term of efficacy with similar progression-free survival (6.9 vs 7.8 months, respectively; HR: 1.00; 95% confidence interval [CI]: 0.82–1.22). Overall risk of cardiotoxicity was significantly higher with doxorubicin than PLD (HR: 3.16; 95% CI: 1.58–6.31; p < 0.001). Overall survival was similar (21 and 22 months for PLD and doxorubicin, respectively; HR: 0.94; 95% CI: 0.74–1.19) [60].
In another Phase III trial comparing the efficacy of PLD with that of a common salvage regimen (vinorelbine or mitomycin-C plus vinblastine) in patients with taxane-refractory advanced breast cancer, progression-free survival and overall survival were similar for PLD and the combination regimen (progression-free survival: HR: 1.26; 95% CI: 0.98–1.62; p = 0.11; median: 2.9 months [PLD] and 0.5 months [combination]; overall survival: HR: 1.05; 95% CI: 0.82–1.33; p = 0.71; median: 11 months [PLD] and 9 months [combination]). In anthracycline-naive patients, progression-free survival was somewhat longer with PLD, relative to the comparator (n = 44; median progression-free survival 5.8 v 2.1 months; HR: 2.40; 95% CI: 1.16–4.95; p = 0.01). The most frequently reported adverse events were nausea (23–31%), vomiting (17–20%) and fatigue (9–20%), and were similar among treatment groups. PLD-treated patients experienced more PPE (37%; 18% grade 3, 0.66% grade 4, one patient only) and stomatitis (22%; 5% grades 3/4). Neuropathy (11%), constipation (16%) and neutropenia (14%) were more common with vinorelbine. Thus, PLD is an effective treatment option for taxane-refractory metastatic breast cancer [61].
Other tumor sites
Preclinical studies in tumor ascites models indicate that there is increased peritoneal accumulation of the liposomal form as opposed to free doxorubicin after intravenous administration [62]. This may explain why intravenous PLD is effective in treating ovarian cancer. PLD has demonstrated efficacy as a single agent in the treatment of recurrent/relapsed ovarian cancer in several clinical trials [63,64]. In a Phase III trial comparing the safety and efficacy of PLD with that of topotecan in 474 patients with relapsed ovarian cancer after first-line, platinum-based chemotherapy, similar overall RRs, median progression-free survival and overall survival were observed. However, in a pre-planned subgroup analysis, patients with platinum-sensitive disease, treated with pegylated liposomal doxorubicin, had a significantly longer median progression-free survival (p = 0.037) and overall survival (p = 0.008) compared with those treated with topotecan [63,65].
Several reports have shown that PLD at a dose of 40 mg/m2 (delivered on an every-4-weeks schedule) the objective RR is similar to that achieved with the US FDA-approved regimen (50mg/m2) (10–15%), but with a substantially improved adverse-effect profile [66–68]. These data provide strong support for the conclusion that, in the palliative setting, the lower-dose treatment program should be employed to optimize the chances for clinical benefit without excessive toxicity. With this data in mind, the National Institute of Clinical Excellence recommends PLD as the first-line therapy in platinum resistant or refractory ovarian cancer.
Apart from breast and ovarian cancer, PLD has been investigated in a variety of solid tumors, such as lung, head and neck, liver and prostate cancers, glioblastoma and multiple myeloma [69–73].
In AIDS-related Kaposi's sarcoma, doxorubicin levels in the lesions were 5.2–11.4-times higher when PLD was used compared with equivalent doses of doxorubicin [15]. This has translated into a more effective treatment clinically when compared with combination chemotherapy with doxorubicin [74].
Conclusion & future perspective
There are several theoretical advantages of using liposomal doxorubicin. These include prolonged half-life, increased drug stability and selective accumulation in tumor, which reduce adverse effects to the heart.
The clinical trials in LAPC and metastatic breast cancer that compared doxorubicin with LEDC at the same dose have showed similar efficacy and better toxicity profile in favor of LEDC. Similar findings of better therapeutic index have been observed with the Phase III trial, compairing 50 mg/m2 of PLD versus 60mg/m2 of doxorubicin, which demonstrated similar efficacy in terms of progression-free survival but much lower cardiac toxicity [60]. The liposomal formulation has a potential advantage for use in combination with trastuzumab, as it has been proved to overcome the major problem of increased cardiac toxicity encountered when doxorubicin is administered concurrently with trastuzumab in breast cancer. Though different single-arm Phase II studies combining PLD and trastuzumab seem reassuring, larger studies are ongoing to further clarify this issue.
Executive summary
The role of doxorubicin in breast cancer is limited by its cardiotoxicity.
Liposomal delivery of doxorubicin prolongs half-life, increases drug stability and tumor site-specific accumulation, thereby reducing cardiac side effects.
Two liposomal doxorubicin formulations are currently used in the treatment of breast cancer: liposomal-encapsulated doxorubicin citrate (Myocet®/D-99) (LEDC), and pegylated (STEALTH®) liposomal doxorubicin (Doxil®/Caelyx®) (PLD).
The reconstitution of LEDC is more complicated than PLD.
LEDC is predominantly taken up from the circulation by the reticuloendothelial system while PLD has prolonged intravascular retention.
The dose limiting toxicity of LEDC is myelosuppression, while that of PLD is mucocutaneous reactions.
Both pegylated and non pegylated liposomal doxorubicin formulations have lower cardiotoxicity than doxorubicin in clinical studies.
Liposomal doxorubicin formulations may be used safely both in anthracycline-naïve and pretreated patients.
Clinical trial results demonstrate that liposomal doxorubicin has similar efficacy and a superior toxicity profile to doxorubicin in breast cancer.
In combination with trastuzumab, PLD has the potential to overcome the problem of increased cardiac toxicity encountered with the doxorubicin and trastuzumab combination. Results of ongoing Phase III trials are awaited.
Higher efficacy of liposomal doxorubicin is observed in some tumors due to better drug delivery, such as ovarian cancer and Kaposi's sarcoma.
Liposomal formulations may be used in the adjuvant setting in breast cancer to reduce long-term toxicity of the drug.
Pharmacoeconomic aspects and emergence of other new drugs with low cardiac toxicity may reduce some of the potential utility of liposomal doxorubicin in combination therapy.
The promising results with liposomal doxorubicin in metastatic breast cancer and LAPC has created a rationale to expand their use in the adjuvant setting, but this should be approached with care, taking in consideration pharmacoeconomic aspects. Though liposomal doxorubicin represents a significant advance in targeted therapy, several other biological agents with impressive efficacy for the treatment of HER-2-overexpressed breast cancer, that are orally administered and also have low cardiac toxicity, are in current clinical development (e.g., the receptor tyrosine kinase inhibitors such as lapatinib). In addition, the long-term safety issues of the liposomal formulation should be carefully evaluated before their routine application for the curative treatment of early-stage disease.
Footnotes
The authors have no relevant financial interests, including employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties related to this manuscript.