
Abstract
This article reviews current understanding of the clinical manifestations, diagnosis and treatment of Sjögren's syndrome. Sjögren's syndrome is a chronic inflammatory disorder of the exocrine glands with multiple nonexocrine features. It is found predominantly in middle-aged women but exists throughout the population. The diagnosis of Sjögren's syndrome can be challenging because the cardinal sicca symptoms may be subclinical or attributed to other causes, such as medications or aging. Differential diagnosis of Sjögren's syndrome can be confounded by the multiple exocrine manifestations in the mouth, eyes, ears, nose, skin, vagina, and respiratory and gastrointestinal tracts, as well as seemingly unrelated nonexocrine involvement in the thyroid, liver, kidneys and the musculoskeletal, vascular and nervous systems. This article concludes that early diagnosis of Sjögren's syndrome is crucial to prevent and/or minimize potentially life-threatening complications. Periodic follow-up of patients’ status and collaboration between the primary-care physician and the rheumatologist, dentist, ophthalmologist and other specialists are indispensable.
Keywords
Sjögren's syndrome (SS) is a chronic inflammatory disease characterized by lymphocytic infiltration of the exocrine glands. Keratoconjunctivitis sicca (KCS) (dry eyes) and xerostomia (dry mouth) are the hallmark of SS. However, the exocrine involvement in SS extends beyond the lacrimal and salivary glands; it includes the entire exocrine system of the gastrointestinal (GI) system, the respiratory system (the nose, sinuses and lungs), the throat, ears, skin and vagina. There are two forms of SS, primary and secondary. In primary SS, the inflammatory process is limited to the exocrine glands, while in secondary SS the exocrine involvement is associated with other connective tissue disease or autoimmune disease, such as rheumatoid arthritis or lupus erythematosus. Both primary and secondary SS may be associated with other, seemingly unrelated, diseases that are often seen in the thyroid, liver, gallbladder, pancreas, kidneys, and the musculoskeletal, vascular and nervous systems [1–9]. Approximately a third of SS patients exhibit systemic nonexocrine involvement [10]. The underlying biochemical and immunological basis for the variation in the clinical presentation is not known. A few studies have suggested that early onset of SS (<35 years of age) is associated with more severe systemic and immunological manifestations than later onset of the disease [11–12] but this distinction in the clinical manifestations between older and younger patients is controversial [13].
Although SS may be mild and subclinical, serious complications, including interstitial pneumonitis, nephritis and hearing loss, may arise [14]. Most noteworthy, perhaps, is that SS patients have a greatly increased (as much as 44-fold) risk for malignant lymphoma [15–17]. In a study of 723 consecutive patients with primary SS, one in five deaths were attributable to lymphoma [18]. In another study of 55 SS cases, Zufferey and colleagues reported that 9% of the patients developed low-grade B-cell lymphoma over a period of 12 years (mean 6.5 years); two of them were in the lymph nodes, one in the parotid, one in the lacrimal, and one in the lungs [19]. They also suggested that patients with systemic manifestations appear to be at higher risk for malignant transformation irrespective of the presence or absence of immunological serum markers such as mixed cryoglobulinemia.
Owing to the broad spectrum of the multisystem involvement in SS, management of the disease often requires a multidisciplinary approach that includes a rheumatologist, dentist, primarycare physician, ophthalmologist, gynecologist, gastroenterologist, pulmonogist, otorhinolaryngologist and dermatologist. This article reviews current understanding of the multisystem nature of the clinical manifestations, diagnosis and treatment of SS.
Prevalence
SS is most prevalent in women in their fourth and fifth decades with a female:male ratio of approximately 9:1 [20]. It is considered the second most common autoimmune disease next to rheumatoid arthritis, with an estimated prevalence of 0.5–5% of the population [21–26]. An estimated 2–4 million individuals in the USA have SS [201], but the majority of them remain undiagnosed. SS may also affect children; there are approximately 140 cases of SS in children in the literature [27–30]. Unlike adult onset, the most common symptom of childhood SS is not sicca symptoms, but recurrent salivary gland infections (mostly parotitis) [31]. It has been suggested that some of the patients who are diagnosed with SS in adulthood might have experienced the early manifestations during their childhood.
Pathogenesis
The exact mechanism(s) of pathogenesis of SS is not known. Despite the relatively common presence of lymphocytes in the exocrine glands of healthy individuals [33], lymphocytic infiltration of the exocrine glands is a characteristic feature of SS and is probably the underlying cause of the damage to the exocrine glands that leads to the loss of their secretory function. However, the lack of correlation between the degree of glandular destruction/focal lymphocytic infiltration and the severity of sicca symptoms suggest that other pathogenic mechanisms play a part. The available research evidence suggests that the pathogenesis of SS is multifactorial and includes immunological, genetic, neurological, infection, environmental and hormonal factors.
It is generally believed that SS is an immune-dysregulation disorder. The elevated levels of serum autoantibodies and B-cell activation in SS support an autoimmune etiology for the disease [20]. The increased serum level and expression of B cell-activating factor (BAFF) in the salivary glands of patients with primary SS are further support for the autoimmune nature of SS [202].
One of the proposed models for the pathogenesis of SS suggests a two-phase process, a non-immune and an immune phase. An exogenous agent could trigger the nonimmune phase. Then the cellular damage and apoptotic byproduct would act as an autoantigens, provoking an immune phase that propagates the inflammatory response [33,34]. Tzioufas and colleagues found a positive association of SS-B antibody with longer disease duration, parotid gland enlargement and a higher proportion of nonexocrine manifestations, compared with patients without autoantibodies [35].
The possibility of immune-mediated neural pathogenesis for SS is supported by the fact that normal lacrimal and salivary function is dependent on neural input to the glands. Cholinergic nerves that release acetylcholine induce tearing and salivation by stimulating muscarinic M3 receptors of the lacrimal and salivary glands [36]. Studies have demonstrated that antibodies from patients with primary SS could bind the M3 receptors of rats' lacrimal glands [37].
The occurrence of SS in individuals with a family history of SS and its association with specific human leukocyte antigen (HLA) suggests a genetic predisposition for the disease [38–41]. Furthermore, it is believed that genetic susceptibility may also be critical for the development of the autoantibody response in SS [35]. There is also an association of specific HLA antigens with the production of SS-A/SS-B autoantibody.
Viral etiology has long been assumed as the potential trigger of the nonimmune phase of the disease. The proposed role of viral infection is believed to be via stimulation of the immune-competent cells that activate the HLA-independent innate immune system, which uses Toll and Toll-like receptors. These receptors recognize conserved molecular patterns (pathogen-associated molecular patterns), which are shared by large groups of microorganisms [42]. Among the viruses that have been implicated in the development of SS are cytomegalovirus, Epstein-Barr virus, T-cell leukemia virus type 1 and HIV [43–47]. Mumps and HIV are known to affect the salivary glands. The inflammatory reaction of HIV-infected glands is not easily distinguishable from that of SS patients. Hepatitis C virus (HCV) clinical cases seem to suggest that this infection is associated with SS too. HCV may propagate and reside within salivary gland tissue, leading to HCV-associated sialadenitis or Sjögren's-like syndrome in some cases. Parotid swelling was reported in a subset of patients with hepatitis C [48] and an overlap of HCV and SS is not uncommon [49–52]. In addition, a direct role for HCV in the physiopathology of certain cases of primary SS has been suggested [47,53].
The high prevalence of SS and other autoimmune rheumatic disorders in middle-aged women and the immunoregulatory properties of androgen, a natural immunosuppressor, and estrogen, an immune stimulator [54,55], suggests a possible role for the sex hormones in the pathogenesis of these conditions. A study from our laboratory suggested that the relative ratios rather than the absolute concentrations of the hormones may contribute to the development and/or progression of SS and could potentially affect the severity of the symptoms [56]. However, the role of sex hormones in SS remains controversial.
Clinical manifestations
Signs and symptoms of SS vary with the nature of glandular and extraglandular involvement. Classically, dry eyes and dry mouth are the most predominant features of primary SS. Other exocrine and nonexocrine manifestations may include the respiratory and GI systems, ears, skin, vagina, kidneys, and vascular, musculoskeletal and nervous systems (
Summary of exocrine and nonexocrine manifestations.
A questionnaire addressing the combined exocrine and nonexocrine symptoms provides a valuable screening tool for SS. Studies have shown that combined symptoms of dry mouth, sore mouth and dry eyes correctly classified 93% of SS patients and 97.7% of the controls. Excluding the mouth, eyes and arthritis, the combined symptoms of nasal, gastric, vaginal, skin, muscle, psychological and thyroid correctly classified 64.3% of SS patients and 86.1% of the controls [57,148]. SS: Sjögren's syndrome.
Respiratory involvement
Although it has been established that respiratory involvement can occur in patients with SS, the prevalence of pulmonary comorbidity is still controversial and has been reported to range from 9–75% [58–62]. This wide variation is in part due to the lack of standardized diagnostic criteria and failure of the studies to consistently differentiate primary and secondary SS.
Nonetheless, SS appears to affect the entire respiratory tract, including the pleura and mediastinum, and has been linked with diffuse interstitial lung disease [63–65], atelectasis and bronchiectasis [66,67], and pulmonary hypertension [68]. In a study of 36 patients with primary SS, the most common respiratory tract finding was diffuse interstitial lung disease (25%), followed by small-airway disease (22%), upper respiratory tract dryness (17%) and obstruction of large airways (8%) [61]. Interstitial pathology seen in patients who underwent transbronchial lung biopsy included dense lymphocytic infiltrates and interstitial fibrosis.
Interstitial lung disease can be categorized as lymphocytic interstitial pneumonitis, pulmonary fibrosis or lymphoma. Also characterized by pulmonary infiltration, pseudolymphoma responds well to corticosteroids, but can advance to lymphoma. Pulmonary fibrosis, present in as many as 10% of patients with primary SS [69], may result from prolonged lymphocytic interstitial infiltration. It was once believed that even secondary SS, as seen in rheumatoid arthritis, put patients at increased risk of bronchiectasis, respiratory infections and airway disease [70–72]. However, it has been suggested that in most patients with secondary SS, respiratory involvement is due to rheumatic disease [73,74].
Loss of mucus secretion in the bronchial tree may result in xerotrachea, chronic bronchitis or small-airway disease. Xerotrachea occurs in approximately a fifth of patients with primary SS [75], and approximately 40–60% of SS patients have hyper-responsive airways as a result of histological abnormalities in bronchi and bronchioles [76,77].
Although often asymptomatic, diffuse lung disease usually presents with dry cough, sometimes characterized as unrelenting, due to xerotrachea [75], or dyspnea; these are common complaints of patients with SS [78].
Objective clinical findings may include crackles heard on auscultation, reticular abnormalities and interstitial-like patterns revealed by chest radiography, and bronchial or peribronchial thickening revealed by computed tomography [1]. Transbronchial biopsies frequently expose bronchiolar lymphoid infiltrates and follicular bronchiolitis [79]. Lymphadenopathy or nodules should be investigated as possible indications of lymphoma [63].
Otic manifestations
The most severe otic manifestation of SS is hearing loss. In a study of 30 patients with SS who underwent audiometric testing, 14 (46%) exhibited sensorineural hearing loss [2]. Hearing loss in patients with SS may be caused by defective production of the permeability barrier of the external acoustic meatus and sensory impairment from nervous system involvement. Otitis externa sicca and fibrotizing external otitis are other complications of SS marked by defects in lamellar body secretion and the production of the permeability barrier [80].
Gastrointestinal & hepatobiliary
Dryness of the pharynx and esophagus often results in dysphagia. However, it is important to exclude other causes of dysphagia such as muscular or neurological diseases and tumors. Chronic atrophic gastritis and lymphocytic infiltrates similar to those in minor salivary glands appear in biopsies of gastric mucosa [1]. Hypopepsinogenemia, elevated serum gastrin levels, low serum vitamin B12 concentrations and antibodies to parietal cells are also seen [81]. Inflammation and destruction of the mucus-secreting glands of the GI tract (Figure 1) and mucosal atrophy have been reported as common findings in patients with primary SS. Mucosal atrophy can affect the entire length of the GI tract and may be associated with upper esophageal webs, dysmotility and achalasia, vasculitis, esophageal candidiasis, and reflux disease. The mechanisms proposed to explain this symptom include the decreased production of saliva, esophageal dysmotility, upper esophageal webs and achalasia [82,83] Mucosal atrophy can also be seen throughout the entire length of the esophagus [82]. Chronic atrophic gastritis commonly occurs in SS [84]. Inflammatory changes in the intestines, related to an immune mechanism, have been described and there is also an increased risk for the development of lymphoma, such as mucosa-associated lymphoid tissue lymphomas within the GI tract [85].
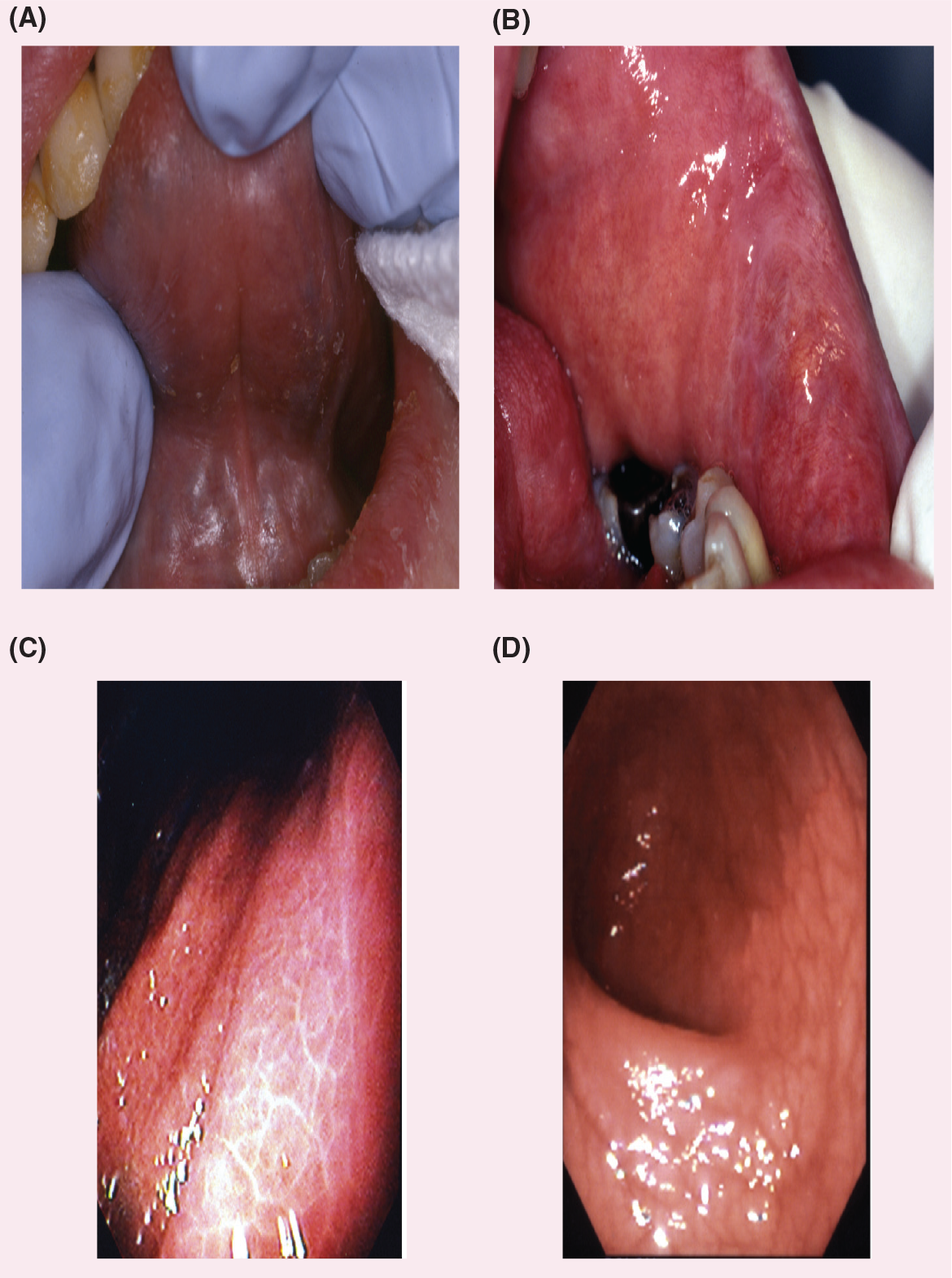
Dry, erythematous mucosa is a predominant feature of Sjögren's syndrome.
Evaluation for malignancy is important if endoscopic studies are performed for symptoms of abdominal fullness or epigastric pain.
Liver involvement was thought to be uncommon (5%) and subclinical, signaling its presence with elevated serum levels of liver enzymes or antimitochondrial antibodies [86]. Biopsy may show features of stage I primary biliary cirrhosis (PBC), and sicca symptoms are seen in 50–80% of these patients [86]. More recently, liver involvement has been found to be a common complication that merits routine investigation in patients with primary SS [8]. In a 5-year follow-up study of patients with SS, Csepregi and colleagues found that high titers of smooth muscle antibodies and anti-mitochondrial antibody can be an indicator for autoimmune hepatic disease and PBC [9]. It has been proposed that the initiating event leading to liver disease starts in the epithelial lining of bile ducts within the liver. Therefore, it is advisable to screen SS patients for these antibodies. It was seen more frequently in patients who also had evidence of lung, kidney and hematological abnormalities. Patients with evidence of liver involvement should be monitored by a hepatologist.
PBC and chronic active hepatitis of the autoimmune type are associated with SS, while HCV infection is currently excluded in classification criteria. A Swedish study of 45 primary SS patients with sicca symptoms found that 27% had abnormal liver enzyme levels [87]. Further evaluation showed that four (9%) of these patients with primary SS also had PBC, and two (4%) had chronic active autoimmune hepatitis.
Chronic pancreatitis and sclerosing cholangitis may result from the eventual fibrosis of the exocrine glands and pancreas in association with the biliary duct system in some cases of SS [88]. Others have reported cases of acute and chronic pancreatitis in association with SS [86–91]. One study demonstrated an immunogenic response to a protein isolated from the pancreas in patients with idiopathic chronic pancreatitis and SS [92]. On radiography or autopsy, pancreatic abnormalities, such as mononuclear-cell infiltration and loss of reticulin pattern, pancreatic calcification and fat necrosis, have been revealed [93–95].
Obstetric & gynecological
There is scant information regarding the effects of SS on the female reproductive tract, and problems potentially related to SS may be missed. Vaginal and vulval dryness are common exocrine manifestations that can result in dyspareunia and pruritus [3]. Unfortunately, like other sicca symptoms, these are often attributed to ‘natural aging’ or menopause. Other pelvic diseases, such as vaginal infection, should be ruled out as the specific cause for dryness is sought.
There are special obstetric considerations for patients with SS. Maternal autoimmune disease results in higher rates of spontaneous abortion and fetal death. Primary SS in the mother has been linked with congenital heart block [96]. Anti-Ro and anti-La antibodies have been shown to cross the placenta and cross-react with the conducting system of the fetal heart [97–99].
Cutaneous & vascular
Reduced exocrine secretions in the skin result in dryness [3]. Heterogeneous skin lesions that are seen include purpura, urticaria, and annular lesions [100,101]. Approximately a third of SS patients also have Raynaud's syndrome, which often long precedes sicca manifestations [1] and disappears or decreases in frequency in approximately half of the affected patients [102]. Vasculitis is limited to the skin in approximately a third of patients with primary SS, appearing as palpable or nonpalpable purpura or lesions similar to urticaria [103] (Figure 2). Leukocytoclastic vasculitis is the most common histological sign. Systemic necrotizing vasculitis, although uncommon, may occur in patients with primary SS. It may occur in association with mixed cryoglobulinemia and takes the form of ulcerative skin lesions, digital gangrene, mononeuritis multiplex, myositis and glomerulonephritis [103]. Occasionally, systemic vasculitis is seen with visceral involvement affecting the kidneys, lungs and GI tract [1] (Figure 2). Patients with secondary SS, which is very often associated with systemic lupus erythematosus (SLE), may exhibit the myriad well-characterized skin manifestations of SLE.

Vasculitis in Sjögren's syndrome can affect the skin and mucosal surfaces.
Musculoskeletal
Secondary SS is also very often associated with rheumatoid arthritis (Figure 3). However, even patients with primary SS who have significant systemic involvement report fatigue, general malaise, low-grade fever, myalgias and arthralgias. They may experience brief episodes of a lupus-like nonerosive arthritis that is sometimes followed by Jaccoud's arthropathy. However, serum levels of muscle enzymes are usually normal or only mildly elevated, unless severe myositis occurs [1]. Arthritis and arthralgia may be present in 60% of patients with primary SS in the form of intermittent polyarticular arthropathy, affecting the small joints, sometimes asymmetrically [103]. Joint deformity and mild erosions are unusual. Myalgias occur frequently in primary SS despite a low incidence of inflammatory muscle disease.

Rheumatoid arthritis (A) and scleroderma (B) in patients with secondary Sjögren's syndrome.
Cardiovascular
Cardiovascular manifestations may include parasympathetic dysfunction. Abnormal heart rate responses to both the Valsalva maneuver and forced respiration were seen in 15% of patients with primary SS in one study, although no signs of sympathetic dysfunction were observed [104]. In a subsequent study using power spectral analysis techniques, investigators found no differences in parasympathetic or sympathetic function between patients with primary SS and healthy controls, although some cardiovascular differences were seen during orthostatic challenge [105].
Isolated congenital heart block has been observed in infants of mothers with primary SS; this was discussed previously in the section on obstetric and gynecological manifestations.
Renal
Renal involvement in SS arises from lymphocytic infiltration of the renal parenchyma or immune complex deposition in the glomeruli. Dysfunction, usually mild and subclinical, is not related to the age of the patient or the duration of disease. Results of the urine acidification test are abnormal in approximately 30% of SS patients, although only approximately 10% have overt renal disease [1,106]. Interstitial disease is the most common renal abnormality, and most of the patients with intertestitial nephritis have hyposthenuria and hypokalemic hyperchloremic distal renal tubular acidosis associated with hypergammaglobulinemia [106]. Distal tubular acidosis can be subclinical or present with recurrent renal colic or hypokalemic muscular weakness. Untreated, it can lead to nephrocalcinosis, renal stones and compromised renal function [107].
Membranous or membranoproliferative immune complex glomerulonephritis associated with cryoglobulinemia and hypocomplementemia has been described in several patients with primary SS [108].
Endocrine
One in two SS patients have subclinical thyroid disease [109], with a greater incidence in primary SS than in secondary SS [110]. The histopathological findings in the thyroid glands of patients with autoimmune thyroiditis resemble those in the salivary and lacrimal glands of patients with SS. It is, therefore, not surprising that autoimmune thyroiditis is the most frequent organ-specific autoimmune disease recognized in patients with primary or secondary SS [111]. Approximately a third of patients with autoimmune thyroiditis are found to have features of SS, indicating that the two diseases may be related pathogenetically [112]. Hypothyroidism may be associated with a hypothalamic deficiency of corticotropin-releasing hormone [113], and some authors have speculated regarding central adrenal deficiency in SS patients. Autoantibodies to thyroid antigens and overt autoimmune thyroid disease are common in SS patients; the syndrome is considered an autoimmune epithelitis that targets polarized epithelium such as that of the thyroid [114]. The prevalence of primary SS among patients with autoimmune thyroiditis was ten-times higher than in the general population, while that of autoimmune thyroiditis was nine-times higher in patients with primary SS than in the general population [115]. In one study of endocrine disorders and immunological parameters in 43 consecutive patients with primary SS, 14 patients (33%) had thyroid disease and 13 (30%) had autoimmune thyroiditis [116].
Furthermore, the hypothalamic–pituitary–adrenal axis, as well as the thyroid axis, has been shown to be hypoactive in SS patients, through assessment of basal and stimulated adrenocorticotropin, cortisol, thyroid-stimulating hormone, and prolactin levels [117]. Hypoactivity of the stress system has been implicated in various systemic autoimmune rheumatic diseases [118,119]. Sex hormones also appear to play an important role in the development of autoimmune disease [120], most likely accounting for the strong female predominance and, in SS, the peak incidence at menopause, when sex hormones fluctuate greatly. Not only is primary SS rare in men, but serological findings in male patients have been reported to differ from those in their female counterparts [121].
Neurological & psychiatric
Disease of the CNS, peripheral nervous system (PNS), or both can develop in patients with SS. The sensory, motor and autonomic components of the PNS may be affected. Peripheral neuropathy is the most common neurological complication of primary SS, occurring in 20–30% of patients [122,123].
The spectrum of PNS disease resembles many types of diabetic neuropathy. Among the most common PNS manifestations of primary SS is neuropathy involving the sensory nerves to the feet and, less often, the hands, which is usually nondisabling. There is also a loss of sensory fibers that recognize position and vibratory impulses essential for maintaining awareness of limb position, posture, balance and normal gait; joint damage may result. Patients may have uncomfortable sensations of pulling or crawling that affect the trunk and extremities. Sensory neuropathy involving the extremities may be accompanied by trigeminal neuralgia. PNS involvement in SS may include carpal tunnel syndrome, caused by the pressure of inflamed tissue against the median nerve [201]. Peripheral sensory or sensorimotor polyneuropathy or mononeuritis multiplex occurs, symmetrically or asymmetrically, in 10–20% of patients with primary SS [1]. Trigeminal neuropathy was the most frequently observed neurological symptom in 21 female patients with primary SS in a Japanese study; it occurred in eight (50%) of the 16 patients with objective neurological abnormalities. CNS involvement was observed in three (14%) of the 21 patients [124].
CNS complications resembling the signs and symptoms of multiple sclerosis (MS) have been reported to occur in as many as 20% of patients with SS [125]. However, later studies in British and Greek populations found no CNS involvement in SS patients, nor was SS present in a sizeable study of MS patients [122,126–127]. A recent report of three cases of SS-associated myelopathy suggested that CNS disease occurs in fewer than 1% of patients with primary SS [128].
Debate regarding the incidence of severe and relapsing neurological disease in SS continues. One study found that most neurological symptoms in patients with primary SS were mild, and that in patients with secondary SS neurological symptoms were more probably caused by the underlying autoimmune rheumatic disease [126]. In another recent study autonomic nervous system dysfunction was no more prevalent in patients with primary SS than in healthy, age- and sex-matched controls [129].
The psychological effects of SS were assessed in a large community-based study in adults aged 18–75 years [22]. Standardized questionnaires evaluated psychological distress, fatigue and health status. SS was defined by serological criteria including presence of at least one of the serum antibodies anti-Ro/SS-A, anti-La/SS-B, antinuclear antibody or rheumatoid factor; unstimulated salivary flow (USF) of 0.5 ml or less in 5 min; positive Schirmer I ocular test (≤5 mm in 5 min); and questionnaire responses regarding ocular and oral symptoms. The syndrome was perceived to have a significant impact on perceived health and well-being, with higher levels of depression and anxiety found in SS patients than in those without the disorder.
Affective disorders developed in a majority of patients with primary SS in a study of neuropsychiatric dysfunction [130]. Patients generally presented with hysteroid dysphoric features. Several patients taking cognitive function tests showed mild memory impairment with attention and concentration deficits. A significant correlation was established between psychiatric disturbances and neurological dysfunction that suggested a possible organic basis for psychiatric dysfunction.
Anxiety, depression and personality structure disorders are frequently seen and possibly associated with dysregulated stress responses [1]. Patients with primary SS scored significantly higher for ‘possible’ clinical anxiety (48%) and depression (32%) on neuropsychiatric questionnaires in comparison with healthy controls [131]. Irritability, low mood, headache, GI symptoms, and impaired concentration and memory were more frequent in patients with primary SS than in those with RA.
Other manifestations
Extreme fatigue may be the most troublesome symptom of SS; many patients spend hours in bed trying to rest or sleep and awake quite tired. Patients with SS rest or sleep on average 2 more hours a day than age-matched healthy peers [132]. Fatigue may also arise from nonrestorative sleep associated with secondary fibromyalgia, depression or multiple awakenings due to dryness. Hypothyroidism, which is frequently associated with SS, although often subclinical, may partially contribute to this. Patients with more severe nonexocrine impairment exhibit easy fatigue and low-grade fever [133].
Fibromyalgia, chronic fatigue syndrome (CFS) and malaise are also detected with SS. Antidepressants, stress reduction and physiotherapy can be useful in managing the symptoms of fibromyalgia, which include musculoskeletal pain, point tenderness and fatigue. Fibromyalgia has been reported with relative frequency in patients with primary SS (22%) [134]. CFS is also associated with extreme fatigue and an array of physical and neuropsychological symptoms that may overlap with those of SS [135]. The differential diagnosis of these conditions is therefore challenging.
Mild normochromic or normocytic anemia is common in SS [1] and an elevated erythrocyte sedimentation rate is detected in approximately 80% of patients with primary SS [3].
Lymphoproliferative disease & mortality
There is an approximately 44-times greater incidence of non-Hodgkin's lymphoma (NHL) among patients with primary SS than among age-matched controls [15–17,136]. One in five deaths in these patients is attributable to NHL [18].
In some patients, there is a progression from benign autoimmune exocrinopathy to pseudolymphoma, and in approximately 5% of patients, to lymphoma [101]. Persistent parotid gland, spleen or lymph-node enlargement is a sign of possible lymphoma. Sudden loss of autoantibodies is also suggestive of malignancy. Lymphoma can begin in the salivary glands, lymph nodes, bone marrow, thymus, liver, spleen, kidney or mucosal sites and localizes in extranodal areas. Malignancy in SS is usually B-cell NHL, but occasionally Waldenström's macroglobulinemia. Whenever there is any suggestion of malignancy, consultation with the appropriate oncologist is indicated.
A long-term, prospective study of the impact of primary SS on morbidity and mortality found that the initial presentation determines the outcome [17]. The overall mortality of patients with primary SS compared with the general population doubled, after adjustment for age and gender, only in patients with adverse predictors. Lymphoproliferative disorders, glomerulonephritis and death were consistently associated with the presence of palpable purpura, low C4 complement levels and mixed monoclonal cryoglobulinemia, the latter two being the more important predictors. Most patients with primary SS did not have adverse predictors and had the same mortality rate as the general population.
Diagnosis
SS is underdiagnosed and often misdiagnosed, primarily due to the diversity of its clinical manifestations, which may resemble or overlap with those of a broad range of autoimmune disorders. In addition, patients do not frequently report dryness symptoms because they consider them part of normal aging, a side effect of medications, a result of mouth breathing, or a result of other causes, but rarely find them related to the onset of another symptom. Physicians, on the other hand, may not inquire regarding dryness symptoms, may consider them insignificant, or may address them individually, thereby overlooking the possibility of a generalized exocrinopathy. Moreover, different definitions and classification criteria that have been used for the diagnosis of SS present an additional challenge to physicians [14]. For these reasons, a delay of up to 10 years between the appearance of initial nonspecific symptoms and clinical diagnosis is not uncommon [1].
US–European classification criteria for Sjögren's syndrome*.
Ocular symptoms: a positive response to at least one of the following questions:
Have you had daily, persistent, troublesome dry eyes for more than 3 months?
Do you have a recurrent sensation of sand or gravel in the eyes?
Do you use tear substitutes more than 3 times a day?
Oral symptoms: a positive response to at least one of the following questions:
Have you had a daily feeling of dry mouth for more than 3 months?
Have you had recurrently or persistently swollen glands as an adult?
Do you frequently drink liquids to aid in swallowing dry food?
Ocular signs: objective evidence of ocular involvement defined as a positive result for at least one of the following two tests:
Schirmer's I test, performed without anesthesia (≤5 mm in 5 min)
Rose Bengal score or other ocular dye score (≥4 according to van Bijsterveld's scoring system)
Histopathology: in minor salivary glands (obtained through normal-appearing mucosa) focal lymphocytic sialoadenitis, evaluated by an expert histopathologist, with a focus score ≥1, defined as a number of lymphocytic foci (which are adjacent to normal-appearing mucous acini and contain more than 50 lymphocytes) per 4 mm2 of glandular tissue
Salivary gland involvement: objective evidence of salivary gland involvement defined by a positive result for at least one of the following diagnostic tests:
Unstimulated whole salivary flow (≤1.5 mL in 15 min)
Parotid sialography showing the presence of diffuse sialectasias (punctate, cavitary or destructive pattern) without evidence of obstruction in the major ducts
Salivary scintigraphy showing delayed uptake, reduced concentration and/or delayed excretion of tracer
Autoantibodies: presence in the serum of the following autoantibodies:
Antibodies to Ro(SSA) or La(SSB) antigens, or both
Adapted with permission from [10].
Although various classification criteria and definitions of SS have been proposed to standardize research, debate continues on their usefulness in clinical practice [10,14,137–139]. As with other rheumatic diseases, there is no single distinguishing hallmark that is diagnostic for SS. The diagnosis of SS requires satisfying a set of clinical and laboratory findings that may not always be present throughout the clinical course of the disease. Due to the importance of early diagnosis for the optimal management of SS, an expert clinician's assessment may still be the ‘gold standard’ to define the diagnosis [10]. Diagnosis requires the presence of subjective symptoms and objective measurement of saliva and tear production, minor salivary gland biopsy, and a number of autoimmune markers (
Management
SS is finally being recognized as a treatable multisystem disorder. Owing to the broad distribution of the exocrine glands and the rheumatological autoimmune nature of the disease, the treatment of SS often requires a multidisciplinary approach that includes, primarily, a rheumatologist for the systemic/rheumatological component, an ophthalmologist for the eyes, and a dentist for the salivary dysfunction and oral complications of the disease. In most cases, consultation with a gastroenterologist, pulmonologist, dermatologist and gynecologist is essential for optimal management of the exocrine component of SS. Frequently, neurological, hematological or oncological consultation is also required.
Treatments are available that can relieve symptoms and limit the damaging local effects of xerostomia and KCS. A regimen of treatment and prevention can go far to reduce the oral and ocular degeneration of chronic xerostomia and KCS.
Two parasympathetic secretagogues are effective in treating sicca symptoms: pilocarpine, a natural alkaloid that stimulates muscarinic receptors in exocrine glands, and cevimeline, a new selective muscarinic agonist that demonstrates high affinity specifically for the M3 receptor located on lacrimal and salivary gland epithelium. By relieving dry mouth, both agents help patients chew, swallow, speak and even sleep more easily. Controlled studies show that cevimeline (Evoxac™) and oral pilocarpine (Salagen®) help improve sicca symptoms and prevent complications [140–143]. For those patients with the most severe salivary dysfunction, who may not benefit from these agents, new treatments including gene therapy and tissue engineering are also being explored.
Revised rules for classification of Sjögren's syndrome.
For primary SS:
In patients without any potentially associated disease, primary SS may be defined as follows:
The presence of any four of the six items indicative of primary SS, as long as either item IV (histopathology) or VI (serology) is positive
The presence of any three of the four objective criteria items (i.e., III, IV, V, VI)
The classification tree procedure represents a valid alternative method for classification, although it should be more properly used in clinical–epidemiological surveys
For secondary SS:
In patients with a potentially associated disease (for instance, another well-defined connective tissue disease), the presence of item I or item II plus any two from among items III, IV, and V may be considered as indicative of secondary SS
Exclusion criteria:
Past head and neck radiation treatment
Hepatitis C infection
AIDS
Pre-existing lymphoma
Sarcoidosis
Graft-versus-host disease
Use of anticholinergic drugs (since a time shorter than fourfold the half-life of the drug)
SS: Sjögren's syndrome. Adapted from [10].
On the horizon are androgen eyedrops to reduce lacrimal gland inflammation (Allergan, Inc., CA, USA), diquafasol tetrasodium eyedrops to activate tear secretion (Inspire Pharmaceuticals, Inc., NC, USA), topical cyclosporine to alleviate the underlying inflammatory process of dry eye (Allergan), and an interferon-α lozenge to stimulate saliva function and improve oral comfort (Amarillo Biosciences, Inc., TX, USA) [203]. Studies are in progress testing the ability of the secretagogues currently used for salivary dryness, cevimeline and pilocarpine, to stimulate tear production. Topical human interferon in lozenge form has shown positive results in short-term studies. However, there is a high placebo response, which may be due to gustatory and mechanical stimulation of the oral mucosa. For systemic disease, tumor necrosis factor inhibitors are being investigated; however, longer-term follow-up is needed to assess the safety and efficacy of biological agents, which appear to have a lesser effect on SS than on rheumatoid arthritis [144,145].
Nonsteroidal anti-inflammatory drugs (NSAIDs) can be helpful for the treatment of painful parotid swelling, if the presence of bacterial infection or lymphoma has been excluded, and for arthritis and arthralgias. Hydroxychloroquine may be used for treatment of arthralgias, myalgias and constitutional symptoms. A small study of hydroxychloroquine showed mitigation of immunological hyper-reactivity (hypergammaglobulinemia and autoantibody levels), but further study of long-term efficacy is needed [146]. A randomized, double-blind, placebo-controlled study of an anti-tumor necrosis factor did not show any evidence of efficacy in the treatment of primary SS [147].
Raynaud's syndrome in primary SS is usually mild and can be managed with protection from cold and calcium channel blockers, such as nifedipine or diltiazem. Mild vasculitis limited to the skin and manifested as purpura usually requires no specific treatment. Occasionally patients may develop systemic necrotizing vasculitis and require steroids, cyclophosphamide and plasmapheresis. Cytotoxic drugs such as cyclophosphamide are reserved for life-threatening systemic manifestations such as necrotizing vasculitis because they significantly increase the risk of lymphoma. The histological type, location and extension guide the treatment of malignant lymphoma.
Treatment of peripheral or CNS disease is accomplished with symptomatic and immunosuppressive agents. Symptomatic medications include aspirin, acetaminophen, NSAIDs, antidepressants and anti-epileptics. Patients can also benefit from supervised exercise, physical therapy and rehabilitation.
No current treatment for peripheral neuropathy addresses either initiation of the vascular inflammatory process or reversal of existing pathology. Immunosuppressive drugs have significant side effects. Recombinant human neurotrophic or growth factors and small molecules that either upregulate expression of neurotrophic factors or mimic them hold great promise for targeted therapy in SS-associated PNS and CNS disease. Neurotrophic factors, such as insulin-like growth factor (IGF)-I, nerve growth factor (NGF) and neurotrophin (NT)3, and new molecules are currently under development as potential treatments for other neuropathies associated with diabetes and chemotherapy.
Immune-regulating (IR) drugs may be of value in treating SS patients and have been shown to downregulate the immunopathological activity of primary SS. It is not clear whether sicca symptoms can be improved. The IR and cytotoxic drugs are primarily used for treatment of severe internal organ involvement, inflammatory vascular disease and malignant B-lymphocyte disease in primary SS. In secondary SS, IR therapy is used against the basic immunoinflammatory connective tissue disease.
Topical moisturizers may be helpful to minimize sicca symptoms. Corticosteroid eardrops can be used to treat the auditory canal and may prevent the need for reconstructive surgery [80]. KCS is treated with artificial tears, eye patching, and boric acid ointment for corneal ulceration. Patients should avoid windy, dry, dusty and smoky conditions as much as feasible [1]. Soft contact lenses may help to protect the cornea, but their use must be monitored carefully due to an increased risk for infection.
Physiological gustatory stimulation of the salivary glands can be helpful for wetting the oral mucosa. Sugar-free candies, lozenges, chewing gum and saliva substitutes can be helpful oral moisturizers [142,144,148]. Prescription fluoride is essential to prevent demineralization and damage to the tooth structure. Although research on salivary gland gene therapy has suggested high potential in animal studies, considerably more research is required before any human clinical testing [149].
For vaginal dryness, nonhormonal vaginal lubricants that are water-soluble, colorless, odorless, nonirritating and residue-free may be useful. Vulval dryness can be managed with a thin application of vitamin E oil, and vaginal yeast infection can be managed with miconazole nitrate cream (Monistat®).
Future perspective
SS remains to be one of the most underdiagnosed conditions because the cardinal sicca symptoms, which are the hallmark of the disease, are frequently attributed to other causes such as medications or aging, leading to delayed diagnosis. Early diagnosis is essential to optimal management, but is often missed due to the potential latency and multiplicity of SS symptoms and their resemblance to those of other disorders. Thus, the expert clinician's assessment may still be the gold standard for diagnosis. The diagnosis of SS requires evaluation of both the exocrine and the nonexocrine components of the disease. A questionnaire addressing the combined exocrine and nonexocrine symptoms provides a valuable screening tool for SS.
Executive summary
Sjögren's syndrome (SS) is a chronic inflammatory disease characterized by lymphocytic infiltration of the exocrine glands. The exocrinopathy in SS is not limited to the salivary and lacrimal glands, it involves the entire exocrine system.
SS is a common inflammatory disease with an estimated prevalence of 0.5–5% of the population and SS patients are at higher risk for malignant lymphoma.
Signs and symptoms of SS vary with the nature of glandular and extraglandular involvement; dry eyes and dry mouth are the most predominant features of the disease. Other exocrine manifestations may include recurrent oral candidiasis and recurrent sinusitis.
The loss of mucus secretion in the bronchial tree may result in xerotrachea, chronic bronchitis and/ or small-airway disease.
Inflammation of the mucus-secreting glands of the gastrointestinal tract and mucosal atrophy are common findings in patients with SS. It can affect the entire length of the gastrointestinal tract and may be associated with esophageal webs, dysmotility, and achalasia.
Peripheral neuropathy is the most common neurological complication of primary SS, occurring in 20–30% of patients.
SS is an underdiagnosed and often misdiagnosed disease, primarily due to the diversity of its clinical manifestations, which may resemble or overlap with a broad range of disorders.
The diagnosis of SS requires satisfying a specific set of criteria, which include the presence of combined clinical and laboratory findings.
Prompt intervention is important to palliate symptoms, prevent and minimize complications, and detect associated disorders. A continuing regimen of prevention and treatment can reduce oral and ocular deterioration, and help ameliorate other exocrine and nonexocrine manifestations.
Patients must be followed regularly for the prevention or early recognition and treatment of complications, related autoimmune diseases and lymphoma. Patients' clinical and laboratory parameters should be monitored periodically.
A multidisciplinary approach including the primary-care physician and the rheumatological, dental, ophthalmological, and gynecological perspectives can play an essential role in early detection and optimal management of this common, underdiagnosed, undertreated, and yet life-altering illness.