
Abstract
Patients with congenital heart disease who wish to become pregnant offer a challenge to obstetricians, cardiologists and anesthetists. Although no large randomized trials exist to support the management of this emerging population, small prospective and retrospective studies provide valuable data on the likely success of pregnancy and the risks involved. Recently, there is emerging consensus on the management of this specialized group of patients, and this article aims to provide the practitioner with an overview of patient needs and the issues to be addressed. All patients with congenital heart disease wishing to consider pregnancy should be referred for specialist assessment prior to conception. Maternal risk, fetal risk and recurrence risk in the fetus should all be addressed. Most women with congenital heart disease can undergo pregnancy without significant risk. However, for some women the risk of maternal death is high, including those with: severe aortic stenosis, impaired left ventricular function, pulmonary hypertension and Marfan syndrome with dilated aortic root. All patients should be offered a detailed 20-week fetal cardiac scan and, in certain cases, prepregnancy genetic counseling. Most patients can deliver vaginally, with cesarean section reserved for obstetric indications or patients in whom straining at delivery could be potentially fatal (i.e., those with Marfan syndrome, aortic aneurysm, severe fixed left heart obstruction, or the acutely unwell mother). Antibiotic prophylaxis should be given routinely in labor to all patients in whom dental prophylaxis is indicated.
As a result of successful pediatric cardiac surgery over the past 30 years, the adult survivors of congenital heart disease will soon outnumber children living with congenital heart disease. In 2000 there were approximately 133,000 adult patients, and this is forecasted to increase to 166,000 by 2010 [1]. Since approximately 50% of the population is female and many are of child-bearing age, adult survivors of congenital heart disease now account for a significant proportion of patients referred in the UK for antenatal cardiac assessment, having superseded patients with rheumatic heart lesions.
Although UK maternal mortality rates are extremely low, being approximately 7.8/100,000 live births, cardiac disease is now the second most common cause of maternal death after psychiatric illness. The majority of cardiac deaths (n = 44), recorded in the last triennial mortality report (2000–02) [2], are secondary to acquired cardiac lesions such as acute myocardial infarction or cardiomyopathy, but congenital heart lesions contribute significantly to maternal morbidity.
Congenital heart disease can be classified as either simple or complex, with complex lesions accounting for approximately 10% of patients. All women with congenital heart disease should be considered for cardiology review prior to conception, and all but the most simple lesions should be referred to a specialist in adult congenital heart disease for further detailed assessment. Such an assessment aims to quantify:
The individual risk of pregnancy to the mother in the short and long term;
The potential risks to the fetus;
The recurrence risk of congenital heart disease in any offspring.
This article aims to provide an overview of the general assessment and management of pregnant patients with congenital heart disease and then focus on the most commonly encountered lesions.
The physiology of normal pregnancy
Before assessing maternal risk, it is useful to first review the physiology of pregnancy to help appreciate why a patient with congenital heart disease may tolerate pregnancy poorly.
Maternal plasma volume steadily increases from early in pregnancy, almost doubling by week 32, and this combines with an approximately 10% increase in heart rate and increased myocardial contractility to increase cardiac output. Cardiac output can almost double, increasing rapidly during the first trimester and reaching its peak by 24 weeks. Both systemic vascular resistance and pulmonary vascular resistance fall. In addition, there is a hormone-induced state of hypercoaguability.
During labor, an increase in sympathetic nervous system activity results in an increase in myocardial contractility, systemic vascular resistance and heart rate, producing a further 15–20% increase in cardiac output and a threefold increase in oxygen consumption.
Blood loss of 500 ml is routine during normal vaginal delivery and approximately 1000 ml during cesarean section, resulting in rapid hemodynamic shifts.
Assessing maternal risk
This can usually be determined from a detailed cardiovascular history and examination, a 12-lead electrocardiogram (ECG), a transthoracic echocardiogram (TTE) and functional assessment employing either a standard treadmill exercise tolerance test (ETT) with pulse oximetry or a metabolic oxygen consumption (MVO2) ETT. Occasionally, further evaluation with cardiac magnetic resonance imaging (MRI) or right and left heart catheterisation studies is required.
A Toronto risk index has been prospectively determined incorporating the following factors, each factor being assigned one point:
A prior cardiac event (heart failure, transient ischemic attack or stroke before pregnancy) or arrhythmia that postdates any corrective or palliative cardiac procedure;
New York Heart Association (NYHA) functional class higher than class II or cyanosis (oxygen saturation less than 90%);
Left heart obstruction (mitral valve area less than 2 cm, aortic valve area less than 1.5 cm or a peak left ventricular outflow tract gradient greater than 30 mmHg by echocardiography);
Reduced systemic ventricular systolic dysfunction (ejection fraction less than 40%).
The estimated risk of a cardiac event (i.e., worsening cardiac functional class, development of overt heart failure, arrhythmias or thromboembolism) in pregnant women with existing cardiac disease with no, one and more than one point is 5, 27 and 75%, respectively [3]. This risk index should be incorporated with lesion-specific risk estimates if available, plus the additional risk posed by anticoagulation. Box 1 outlines the conditions in which pregnancy can be categorized as low, moderate or high risk.
Maternal risk may be minimized in certain circumstances by performing a timely catheter or surgical intervention prior to conception. The cardiac status of some women with congenital heart disease will inevitably deteriorate with time, resulting in an increased risk during pregnancy. Such women should be advised that pregnancy would be better tolerated in their twenties rather than late thirties.
Assessing fetal risk
The risk of a fetal complication i.e., premature birth, small birthweight for gestational age, respiratory distress syndrome, intraventricular hemorrhage and fetal/neonatal death, is greatest in women aged less than 20 years or over 35 years, and in those who smoke, receive anticoagulants or have multiple gestations [4]. Those with one or more cardiac risk factors (i.e., left heart obstruction, poor functional class, or cyanosis) have an additional risk of fetal complications.
Potentially teratogenic cardiac medications, such as ACE inhibitors, are generally discontinued once pregnancy is confirmed, but anticoagulation therapy cannot usually be safely stopped. Warfarin crosses the placenta with a risk of teratogenic effects in the first trimester and of fetal hemorrhage throughout pregnancy. In most instances, therefore, heparin (which does not cross the placenta and has no fetal effects) is substituted for warfarin for the duration of pregnancy. However if the mother has a mechanical valve, the interests of the mother and fetus are in conflict. In this situation, heparin is a less effective anticoagulant than warfarin, putting the mother at risk of potentially fatal valve thrombosis. Both patient and doctor face a dilemma as to the safest anticoagulation regimen.
Recurrence risk in the fetus
The risk of congenital heart disease in the general population is less than 0.6%. However, if a first-degree relative is affected this risk can increase tenfold, with the recurrence risk in the fetus being greatest when the mother is affected. Left-sided obstructive lesions are known to have an increased risk of transmission. Certain lesions known to have autosomal dominant inheritance, such as Marfan syndrome or the 22q11 deletion syndrome, confer a 50% risk of inheritance in the offspring [5]. Patients should, therefore, be offered prepregnancy genetic counseling and a detailed 20-week fetal cardiac scan.
Assessment of lesion-specific risk.
Uncomplicated, small or mild:
Pulmonary stenosis
Ventricular septal defect
Patent ductus arteriosus
Mitral valve prolapse with no more than trivial mitral regurgitation
Successfully repaired simple lesions:
Ostium secundum atrial septal defect
Ventricular septal defect
Patent ductus arteriosus, Total anomalous pulmonary venous drainage
Native valve disease excluding severe left-sided stenotic lesions
Mechanical valves
Mild left-ventricular impairment
Repaired coarctation of the aortic
Marfan syndrome without aortic dilatation or family history of dissection
Repaired tetralogy of Fallot
Systemic right ventricle (with moderate/ good systolic function)
Fontan-type circulation (with good left ventricular function)
Cyanotic heart disease with normal pulmonary arterial pressures
Severe left ventricular outflow tract obstruction (Echocardiogram-derived mean gradient >50 mmHg, or peak gradient >80 mmHg)
Severe mitral stenosis
Marfan syndrome with aorta dilated more than 40 mm
NYHA functional class III/IV or systemic ventricular ejection fraction <30%
Eisenmenger syndrome and pulmonary arterial hypertension of any cause.
The conditions in this category cover a broad range of risk depending on the risk index described in the text. Risks are additive, so these conditions may become high risk if more than one risk factor is present. NYHA: New York Heart Association.
Management during pregnancy
Once the risks have been established, the pregnant woman should ideally be followed in a joint cardiac/obstetric clinic. Occasionally a patient may need admission for bed rest from the second trimester onwards.
An individualized delivery plan should be formalized well in advance of the due date after consultation between the patient, her obstetrician, her cardiologist and the cardiac obstetric anesthetist. In general, all but the simplest lesions should be delivered at the regional centre. Most patients should be allowed to go to term, with induction reserved for obstetric indications and those who begin to decompen-sate in the third trimester. Invasive cardiac monitoring is sometimes needed, with an intra-arterial line and central venous line usually sufficient for hemodynamic assessment of complex patients. Air filters should be used in all patients with a right-to-left shunt in order to prevent paradoxical embolism.
A normal vaginal delivery is the aim, with cesarean section being reserved for obstetric indications or patients in whom straining at delivery could be potentially fatal, such as those with Marfan syndrome, aortic aneurysm, severe fixed left heart obstruction, or an acutely unwell mother. Adequate analgesia is important and slow-onset epidural anesthesia can be safely used in most conditions. The left lateral position may need to be adopted by the patient during labor to reduce compression of the inferior vena cava by the gravid uterus and, if necessary, the second stage of labor (i.e., delivery of the fetus) can be shortened with either forceps or vacuum assistance in order to minimize hemodynamic fluctuations. Although American Heart Association (AHA) guidelines only recommend antibiotic prophylaxis for complicated deliveries of patients in whom dental antibiotic prophylaxis is recommended, it is routinely given to all patients in many UK centers at onset of labor, since antibiotics are likely to be forgotten if an unexpected complication arises during labor and delivery.
Patients deemed to be at intermediate or high risk should be carefully monitored for 72 h postpartum since the altered hemodynamics take many days to normalize and may indeed take many weeks to fully return to prepregnancy levels [6].
Common simple lesions
Congenital pulmonary stenosis
Pulmonary stenosis is usually an isolated lesion and severity can be assessed prepregnancy according to peak transvalvar gradient derived by echocardiography (i.e., trivial: <25 mmHg; mild: 25–49 mmHg; moderate: 50–79 mmHg; severe: >80 mmHg).
Mild and moderate pulmonary stenosis or previously treated lesions are usually well tolerated in pregnancy, providing right-ventricular function is good. Those patients with severe stenosis, even if asymptomatic, should be offered balloon valvuloplasty or surgery preconception, since the increased hemodynamic load can precipitate heart failure and/or atrial arrhythmias. If cardiac compromise occurs during pregnancy, balloon valvuloplasty can be offered, preferably late in the second trimester.
Left ventricular outflow tract obstruction
Bicuspid aortic valvar stenosis is the most commonly encountered lesion; however, fixed supravalvar and subvalvar lesions will behave similarly. As the stroke volume increases and peripheral vascular resistance falls, the outflow tract gradient increases. Thus, the baseline gradient should be assessed prepregnancy. Those patients who are asymptomatic, with a peak echo gradient under 80 mmHg, mean gradient below 50 mmHg, with good left-ventricular function, with no ST segment depression on ECG and who have a normal exercise test (with an appropriate rise in blood pressure and heart rate), are likely to tolerate pregnancy. Symptomatic patients and those with severe left-ventricular outflow tract obstruction should be offered surgical relief of the obstruction prior to conception.
Decompensation during pregnancy, suggested by a decreasing gradient due to deterioration in left-ventricular function, can be treated symptomatically with oxygen, diuretics and β-blockers. The aim is to reach a stage in the third trimester when the baby can be safely delivered. Urgent balloon valvuloplasty can be offered in order to delay delivery. Emergency valve replacement is seen as a last resort since it carries a 30% risk of fetal loss and an 11% operative maternal mortality [7].
Left-to-right shunts
This encompasses patients with atrial septal defect (ASD), ventricular septal defect (VSD) and patent ductus arteriosus (PDA). Patients with previously repaired lesions with no evidence of pulmonary hypertension, normal ventricular function and no hemodynamic sequelae are at no increased risk as compared with a pregnant woman without heart disease [8]. Those with unoperated lesions and no pulmonary hypertension or ventricular dysfunction normally tolerate pregnancy well, as the increased cardiac output and hence potential volume overload is offset by the decrease in peripheral vascular resistance. There is a risk of arrhythmia and ventricular dysfunction, especially in those with a large left-to-right shunt. Endocarditis prophylaxis is generally recommended for all patients with structural heart disease, although for ASD this is unnecessary.
In those patients with ASD there exists the potential for reversed shunting resulting in paradoxical embolism, especially during delivery. Therefore, intravenous lines should be avoided, and if used, combined with an air filter. Immobile patients should be treated with graded compression stockings and low molecular-weight heparin prophylaxis.
Coarctation of the aorta
Coarctation of the aorta is a narrowing of the aorta distal to the left subclavian artery, which is frequently associated with hypoplasia of the aortic arch and abnormalities of the head and neck vessels. Most pregnant women will have previously repaired lesions and MRI should be used to assess for recoarctation and aneurysm formation at the repair site and reoperation performed prior to conception. All patients should undergo regular blood pressure measurement and echocardiogram assessment of the coarctation site. Those patients with hypertension or aneurysm should be treated with β-blockers throughout pregnancy. If there is concern regarding aneurysm formation/expansion during pregnancy, MRI can be safely undertaken with the patient lying in the left lateral position or supine with a pelvic wedge. Vaginal delivery with an assisted second stage is recommended except in the presence of an aneurysm, when cesarean section is indicated.
If the diagnosis of coarctation is made during pregnancy, aggressive blood pressure management is required with elective cesarean section at approximately 35 weeks.
The major maternal risk, whether repaired or unrepaired, is dissection of the aorta, usually occurring in the third trimester or during labor. Fortunately, maternal death is rare in both groups [9].
Marfan syndrome
Marfan syndrome is an inherited connective tissue disorder, which classically presents with skeletal, cardiovascular and ocular manifestations. It occurs due to mutations in the gene encoding fibrillin-1, an important component of the extracellular matrix microfibril.
When assessing a potential mother with Marfan syndrome there are two important issues to be addressed. First, since it is an autosomal dominant condition there is a 50% risk of transmission to the fetus. The mother needs to be offered counseling and possible fetal karyotyping if a mutation has been identified in an affected parent.
Second, cardiac involvement occurs in approximately 80% of patients and this determines prognosis. Classically, there is dilatation of the aortic root due to an underlying medial aor-topathy. Progressive aortic dilatation may occur in pregnancy as a result of the increased hemodynamic stress and possibly secondary to hormonal effects with the consequent risk of aortic dissection and valvar regurgitation. Aortic dissection carries a 1% risk of maternal death and a 22% risk of fetal death [10].
The risk of dissection is related to the diameter of the aortic root, however, the size warranting preconception referral for elective aortic root replacement or advice to terminate a pregnancy is open to debate. The European guidelines recommend a diameter greater than 40 mm, quoting a 10% risk of dissection at this size compared with 1% if the diameter is less than 40 mm [11], whereas the Canadian guidelines recommend a diameter over 44 mm [12]. A recent prospective study assessing the aortic root of 22 women during 31 pregnancies concluded that pregnancy appeared to be relatively safe up to an aortic root diameter of 45 mm [13]. A history of a rapidly increasing aortic root (>10 mm per year) or a family history of dissection are additional risk factors to be considered.
All patients, including those who have previously undergone aortic-root replacement, should receive β-blocker therapy throughout pregnancy, and the aortic root should be regularly assessed by echocardiography. Evidence of a rapidly expanding aortic root would necessitate emergency root repair/replacement, which carries a 22% risk of maternal death in pregnancy and the puerperium [7].
Pregnancy is usually well tolerated in patients with a normal aortic-root dimension or in those who have undergone previous aortic root replacement and have no other evidence of cardiac involvement. However, it still carries a potential risk of aortic dissection and the patient needs to be advised accordingly.
Complex lesions
Tetralogy of Fallot
Tetralogy of Fallot, probably the most commonly encountered complex lesion, comprises a ventricular septal defect (VSD), above which the aorta overrides, subpulmonary/pulmonary valvar obstruction and consequent right-ventricular hypertrophy. Most patients considering pregnancy will have undergone radical repair in infancy, therefore, the major concerns are the degree of residual subpulmonary/pulmonary obstruction, the severity of the existing pulmonary regurgitation (occurring as a result of previous transannular pulmonary repair), any persistent VSD and an assessment of the right-ventricular function. Patients with mild impairment of right-ventricular function, with even severe pulmonary regurgitation usually tolerate pregnancy, providing they are asymptomatic prior to conception. They may, however, become breathless during the later stages, requiring bed rest and diuretic therapy. Those who are symptomatic or have significant right-ventricular impairment as a consequence of right-ventricular outflow tract obstruction or pulmonary regurgitation require corrective surgery prior to undertaking pregnancy. In the largest study of the outcomes of pregnancy in tetralogy of Fallot there were 50 successful pregnancies in 26 patients, with 19% suffering a complication (symptomatic heart failure, arrhythmias or both).
Patients who are asymptomatic with severe pulmonary regurgitation and good right-ventricular function are unlikely to develop significant complications during pregnancy. Those with severe pulmonary regurgitation who are symptomatic or have significantly impaired right-ventricular function should be considered for pulmonary valve replacement prior to conception [14,15].
All patients with tetralogy of Fallot should be offered genetic counseling prior to conception with assessment for the 22q11 deletion syndrome. In its absence, the risk of defects in the fetus is 4–6% [11].
Transposition of the great arteries
Transposition of the great arteries (TGA) describes the condition whereby the aorta arises from the right ventricle and the pulmonary artery from the left ventricle. Shunting at atrial level and maintenance of the ductus arteriosus enables initial survival of the neonate. Until the 1980s, repair was most commonly by rerouting the blood at atrial level to the correct great vessel occurring by means of an atrial baffle, known as either a Senning or Mustard procedure (Figure 1). The important consequence of this is the right ventricle becomes the systemic ventricle and hence has to cope with a high pressure circuit, unlike the left ventricle which simply pumps blood to the lungs. Therefore, the main concerns for a woman contemplating pregnancy are:
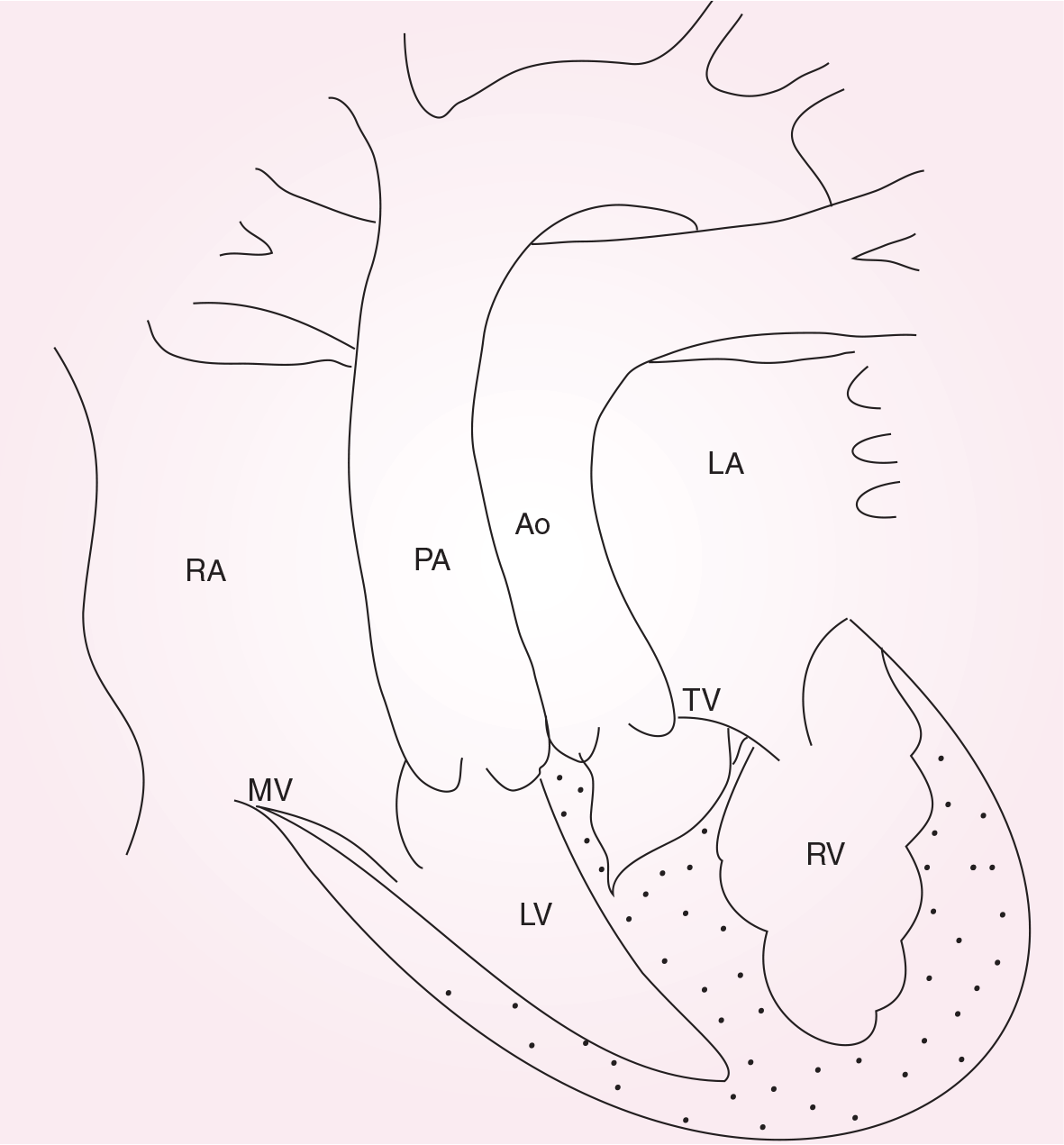
Schematic representation of congenitally corrected transposition of the great arteries.
The atrial baffle, which can either leak or become obstructed. Pulmonary venous pathway obstruction is particularly poorly tolerated and should be corrected prior to conception;
The pulmonary arterial pressure;
The function of the systemic (right) ventricle;
The severity of any systemic atrioventricular valve (tricuspid valve) regurgitation.
Transoesophageal echo or cardiac MRI may be needed for adequate preconception evaluation.
A patient in NYHA functional class I or II with an unobstructed atrial pathway, normal pulmonary pressures, good systemic ventricular function and less than moderate tricuspid regurgitation should tolerate pregnancy, although there is a risk of atrial arrhythmia or sinus-node dysfunction (consequent upon previous atrial surgery) and worsening right-ventricular function as pregnancy progresses. Regular clinical and echo assessment is warranted with a low threshold for admission for bed rest and diuretic therapy. In the largest study to date assessing outcome of pregnancy after atrial repair, 28 women had 69 pregnancies, which resulted in 71% live births. The most important cardiac complication was significant arrhythmia occurring in 22% of women [16].
Congenitally corrected transposition of the great arteries
Congenitally corrected TGA (Figure 2) describes a condition whereby both the atrioventricular and ventriculoarterial connections are discordant, which results in the right ventricle becoming the high-pressure systemic ventricle and the left ventricle supplying blood to the pulmonary circuit. Providing there are no associated cardiac lesions, no surgery is indicated in infancy and a patient can often remain undetected until complications develop in adult life. The principles governing management in pregnancy are similar to those of a patient with TGA, i.e., a patient in NYHA class I or II with normal pulmonary arterial pressures, good systemic ventricle and less than moderate tricuspid regurgitation should tolerate pregnancy. This was demonstrated in the most recent study describing 60 pregnancies in 22 women, with only one woman developing heart failure late in pregnancy secondary to systemic (tricuspid) AV valve regurgitation [17].

Schematic representation of transposition of the great arteries post atrial repair (Mustard or Senning operation).
Fontan-type circulation
This is a surgically created circuit that directs blood from the superior and inferior vena cavae directly into the pulmonary arteries, bypassing the right ventricle and in many cases the right atrium (Figure 3). It is a palliative procedure devised in the mid-1970s and modified over later years for the treatment of patients with a functionally univentricular heart, for example, tricuspid atresia, pulmonary atresia with intact ventricular septum or double inlet ventricle.

Schematic representation of Fontan-type operation (total cavopulmonary connection) for tricuspid artesia.
For a Fontan-type circulation to function effectively the pulmonary vascular resistance must remain low and the left atrial pressure must be low; the latter is dependent upon adequate left-ventricular function. The circuit is extremely vulnerable to atrial arrhythmia and thrombosis and there is limited ability to increase cardiac output.
Prior to pregnancy it is vital to assess the Fontan circuit either by transesophageal echocardiogram or cardiac MRI to ensure that there is no obstruction to either systemic venous return or pulmonary venous return and that left-ventricular function is acceptable (ejection fraction >40%). If this is satisfactory, then providing the patient is in NYHA functional class I or II and performs well with exercise testing, then pregnancy can be considered providing that the patients is aware of the increased risks of atrial arrhythmia, left-ventricular failure and thrombosis of the Fontan circuit [18]. All adults with a Fontan circuit should probably be on lifelong anticoagulation with warfarin and this should be converted to low molecular-weight heparin for the duration of pregnancy. During delivery it is vital to maintain adequate filling pressures and avoid dehydration and vasodilatation.
Cyanotic heart disease with normal pulmonary pressures
Any cardiac lesion that allows right-to-left shunting of blood will result in maternal hypoxemia and hence, central cyanosis. Such conditions include unoperated or palliated complex cyanotic conditions with well-balanced single ventricle circulations. The additional problems encountered with cyanotic patients are:
An increased level of cyanosis during pregnancy due to the combination of reduced systemic vascular resistance and increased cardiac output exacerbating right-to-left shunting;
A risk of paradoxical embolism, especially during labor;
An enhanced thrombotic risk due to secondary erythrocytosis triggered by hypoxemia;
An increased risk of hemorrhage due to decreased vitamin K-dependent clotting factors, thrombocytopenia and platelet malfunction;
Heart failure precipitated by the additional increase in cardiac output on an already volume-overloaded ventricle.
The largest study to date assessing 96 pregnancies in 44 women with cyanotic congenital heart disease (Eisenmengers syndrome excluded) demonstrated a maternal morbidity of 32% and one maternal death postpartum [19].
Management of such patients includes limitation of exercise, supplemental oxygen and graded compression stockings. Intravenous lines should be fitted with an air filter to prevent air embolism. Patients should be well hydrated and vasodilator therapy, such as high-dose epidural anesthesia, should be avoided since it can result in increased right-to-left shunting. Vaginal delivery is the mode of choice, although fetal problems frequently result in the need for cesarean section.
The main concern to the fetus is the effect of increasing maternal hypoxia, which causes intrauterine growth retardation, resulting in low birthweight, prematurity and an increased risk of first-trimester spontaneous abortion. Fetal viability is related to the degree of maternal hypoxia, with 12% live births occurring with maternal oxygen saturations of less than 85% compared with 92% live births at greater than 90% saturation [19].
The Eisenmenger syndrome
Eisenmenger syndrome is defined as pulmonary hypertension (>75% systemic) with reversed or bidirectional shunting occurring at any level, i.e., atrial ventricular or pulmonary. Such patients are cyanotic and as such experience the attendant complications previously described. However, it is the degree of pulmonary hypertension that puts these patients at extremely high risk, since the poorly compliant pulmonary vasculature and impaired right-ventricular reserve are unable to adapt to the increases in cardiac output and oxygen demand, resulting in a maternal mortality in the order of 40–50%, a figure that has remained unchanged over the past four decades [20].
Patients with Eisenmenger syndrome should be counseled regarding the high risk of pregnancy from an early age and offered reliable contraception with either a progestogen subdermal implant (Implanon™, Organon, NJ, USA) or laparoscopic sterilization, (although the latter is not without risk). Patients should be advised that reliable contraception is paramount, since even termination of an unwanted pregnancy carries significant risk. If pregnancy does occur, patients may require admission for bed rest from the second trimester onwards and general management should be as for any cyanotic patient. Therapeutic agents aimed at preferential pulmonary vasodilatation have been used, for example, nifedipine, prostacyclin, nitric oxide and sildenafil, although there is no evidence to suggest that any intervention improves outcome. During labor, intra-arterial monitoring should be used to monitor blood pressure and oxygen saturation. Since the majority of deaths occur postpartum [20], the mother should be monitored for at least 2 weeks in hospital following delivery.
Mild pulmonary hypertension
Pulmonary hypertension (a mean pulmonary artery pressure ≥25 mmHg at rest, or 30 mmHg with exercise), carries a significant risk of maternal mortality. Importantly, even mild pulmonary hypertension can progress during pregnancy, therefore every patient needs to be fully assessed and advised accordingly.
Conclusion
Early recognition of patients with congenital heart disease and a multidisciplinary approach to the management of these complex patients, combined with the continued education of patients and practitioners alike, should aid in the desired outcome of a live baby and well mother. As the population of adult survivors of congenital heart disease grows, so too will the need for trained professionals to meet their needs in this fascinating area of medicine.
Future perspective
Over the next 5–10 years, as the population of patients born with congenital heart disease continues to expand, experience in the management of pregnancy in such patients will increase. It is hoped that larger scale prospective studies will emerge, providing further data on the management of this challenging population of patients. Combined specialist obstetric and cardiology clinics must become the standard of care in the follow-up of these women, integrated with anesthetic and genetic services.
In addition, new developments in pediatric cardiac surgery will result in an ever-changing patient population, and patients who have undergone pioneering palliative procedures, such as the Norwood procedure for hypoplastic left heart, will be reaching adulthood and may wish to contemplate pregnancy. Hence, there is no room for complacency in this continually evolving area of medicine, as more questions will be posed requiring more and more answers.
Executive summary
There is a growing population of adult survivors of congenital heart disease well enough to contemplate pregnancy.
Cardiac disease is the commonest cause of maternal death in the UK.
All women with congenital heart disease should be referred for appropriate expert assessment and counseling prior to conception.
Major cardiovascular changes occur during pregnancy.
Prepregnancy assessment should consider the ability of the congenitally abnormal heart to adapt to the physiological demands of pregnancy and delivery.
Maternal risk can be assessed with: Appropriate use of prepregnancy investigations; Use of the Toronto risk score; Consideration of lesion specific risks.
Where possible, maternal risk should be minimized by intervention or surgery prior to conception.
Risks are additive.
The fetus is at risk of: Fetal or neonatal death; Prematurity; Being small for gestational age; Recurrence of congenital heart disease.
Maternal factors which increase cardiac risk: Noncardiac; Cardiac, including cyanosis, poor functional class; Teratogenic medication.
Joint care by a specialist team of a cardiologist, obstetrician, anesthetist, midwife and congenital cardiac nurse specialist is the key to high-quality management.
An individualized antenatal, delivery and postpartum plan should be made.
Vaginal delivery is the safest mode of delivery for most women with heart disease.
The altered hemodynamics of pregnancy take at least 6 weeks to return to normal.
The management of pregnancy in some specific common simple (but not necessarily low risk) conditions is considered: pulmonary stenosis, left ventricular outflow tract obstruction, left to right shunts, coarctation of the aorta, Marfan syndrome.
The management of pregnany in some specific complex conditions is considered: Tetralogy of Fallot, transposition of the great arteries, congenitally corrected transposition of the great arteries, Fontan circulation, cyanotic heart disease, Eisenmenger syndrome, mild pulmonary hypertension
Executive summary
More appropriately trained professionals are required to meet the needs of this growing population of survivors.
There is a need for large-scale prospective studies of pregnancy risk assessment, management and outcome and in this population.
Combined specialist obstetric and cardiac clinics should become the norm.
Innovations in pediatric heart surgery result in an ever-changing population of young adults with increasingly complex hearts. Their needs will continue to challenge and reward the professionals involved in their care.