
Abstract
Increasing evidence indicates that reactive oxygen species (ROS), consisting of superoxide, hydrogen peroxide, and multiple others, do not only cause oxidative stress, but rather may function as signaling molecules that promote health by preventing or delaying a number of chronic diseases, and ultimately extend lifespan. While high levels of ROS are generally accepted to cause cellular damage and to promote aging, low levels of these may rather improve systemic defense mechanisms by inducing an adaptive response. This concept has been named mitochondrial hormesis or mitohormesis. We here evaluate and summarize more than 500 publications from current literature regarding such ROS-mediated low-dose signaling events, including calorie restriction, hypoxia, temperature stress, and physical activity, as well as signaling events downstream of insulin/IGF-1 receptors, AMP-dependent kinase (AMPK), target-of-rapamycin (TOR), and lastly sirtuins to culminate in control of proteostasis, unfolded protein response (UPR), stem cell maintenance and stress resistance. Additionally, consequences of interfering with such ROS signals by pharmacological or natural compounds are being discussed, concluding that particularly antioxidants are useless or even harmful.
1. INTRODUCTION
Mitochondrial metabolism and reactive oxygen species
Mitochondria are important cell organelles that are responsible not only for the conversion of the bulk of nutritive energy, but also exert a major role in aging processes and in the development of age-related diseases. As an inevitable by-product of oxidative phosphorylation (OxPhos), mitochondria generate over 90% of all intracellular reactive oxygen species (ROS), with conversion of 0.15 − 5 % of total oxygen consumed by resting cells (Halliwell and Gutteridge 2007, Chance et al. 1979, Boveris and Chance 1973, St. Pierre et al. 2002). Hence, as main producers of energy and also potentially harmful ROS, mitochondria have a major impact on physiological and pathophysiological processes within the cell.
ROS formation to an extent that exceeds physiological levels and hence causes putative damage is called oxidative stress (Sies 1985). Thus, mitochondrial dysfunction implicating increased oxidative stress has been proposed to be associated with a variety of diseases like diabetes, cancer and neurodegenerative disorders, including Alzheimer's and Parkinson's disease (Wiederkehr and Wollheim 2006, Ristow 2006, Fukui and Moraes 2008, Tatsuta and Langer 2008). Beyond that, impairment of mitochondrial activity is supposed to be a major reason for aging (Tatsuta and Langer 2008, Bratic and Larsson 2013, Trifunovic et al. 2004), whereas the role of ROS in this regard is still under debate. On the one hand, ROS have been implicated into cellular damage hence contributing to the aging process. On the other hand, an increasing number of studies linking improvement of mitochondrial capacity to increased lifespan and health span extension. Evidence for this dates back to the 1990s, when essential signaling roles for hydrogen peroxide were established (Barja 1993, Finkel 1998, Sena and Chandel 2012). Hence, it seems that a shift towards oxidative metabolism could delay the onset of age-related diseases and maybe aging itself.
Free radical theories of aging
Increased formation of mitochondrial ROS was postulated to be a major cause of aging in 1956, when Denham Harman introduces his Free Radical Theory of Aging (FRTA) (Harman 1956). According to this concept, increased ROS formation causes an accumulation of damage in the cell within age, resulting in age-related impairment of cellular functions and ultimately death of the cell or the corresponding organism, respectively. Respiratory enzymes, which utilize oxygen to generate readily available energy, were proposed to be the main generators of ROS. Due to the fact that mitochondria are the main intracellular source of ROS, Harman extended his initial FRTA theory to the Mitochondrial Free Radical Theory of Aging (MFRTA) (Harman 1972). Over the last decades, significant research efforts have been invested to prove the MFRTA, however generating inconsistent and conflicting results (Perez et al. 2009). Accordingly, nowadays it seems to be established that enhancement of metabolic rate does not necessarily result in concomitantly increased ROS formation (Lapointe and Hekimi 2010) and that the relationship between ROS levels and aging is not linear (Delaney et al. 2013, Johnson et al. 2001, Lee et al. 2003, Kim and Sun 2007, de Castro et al. 2004). Nevertheless and supporting the MFRTA, a bulk of studies in different organisms found that reduced levels of oxidative stress result in extended lifespan (Harrington and Harley 1988, Phillips et al. 1989, Orr and Sohal 1994, Parkes et al. 1998, Melov et al. 2000, Moskovitz et al. 2001, Bakaev and Lyudmila 2002, Ruan et al. 2002, Ishii et al. 2004, Huang et al. 2006, Zou et al. 2007, Kim et al. 2008, Quick et al. 2008, Dai et al. 2009, Shibamura et al. 2009) and long-lived species seem to produce fewer ROS and accumulate less damage than short-lived organisms (Gredilla et al. 2001, Sanz et al. 2010, Sanz and Stefanatos 2008, Gruber et al. 2008).
As a consequence, ROS-lowering interventions were widely proposed to be a promising strategy to retard aging in humans. In this regard, natural or artificial substances that are able to scavenge ROS, so-called antioxidants, were examined intensively. In contrast to the studies in lower model organisms cited above, several prospective intervention trials did not find any health-promoting effects of antioxidant supplementation. Unexpectedly, most interventional studies found a lack of effects in humans (Greenberg et al. 1994, Liu et al. 1999, Rautalahti et al. 1999, Virtamo et al. 2000, Various 2002, Sacco et al. 2003, Zureik et al. 2004, Czernichow et al. 2005, Czernichow et al. 2006, Cook et al. 2007, Kataja-Tuomola et al. 2008, Sesso et al. 2008, Katsiki and Manes 2009, Lin et al. 2009, Song et al. 2009), whereas others even suggested detrimental effects on human health, for instance promotion of cancer growth or induction of diseases with negative impact on human lifespan (Albanes et al. 1996, Omenn et al. 1996, Vivekananthan et al. 2003, Lonn et al. 2005, Bjelakovic et al. 2007, Ward et al. 2007, Lippman et al. 2009, Schipper 2004, DeNicola et al. 2011, Abner et al. 2011). Consistently, several studies overexpressing antioxidant enzymes in mice failed to exert positive effects on lifespan or associated parameters (Jang et al. 2009, Muller et al. 2007, Perez et al. 2011). Accordingly, several long-lived species were found to have a relatively lower expression of antioxidant genes than short-lived ones (Brown and Stuart 2007, Lopez-Torres et al. 1993, Page et al. 2010, Page and Stuart 2012, Salway et al. 2011). In the fruit fly Drosophila melanogaster, increasing levels of mitochondria-derived ROS were found during aging, but were not altered through interventions that increase longevity (Cocheme et al. 2011). Finally, mice that are heterozygous for Mclk1, coding for a ubiquinone synthesis enzyme, showed both increased mitochondrial ROS production and extended lifespan (Liu et al. 2005).
Mitochondrial hormesis (Mitohormesis): Non-linear responses to increased levels of ROS
These before-mentioned findings fundamentally questioned the FRTA, eventually requiring a modernized view concerning the putative roles of mitochondrial ROS (mtROS) generation. It has been repeatedly shown in recent years that mtROS serve as important signaling molecules mediating both cellular and systemic physiological changes, which has been summarized elsewhere (Finkel 1998, Mittler et al. 2011, Sena and Chandel 2012). Physiological targets for ROS are, for instance, thiol groups on cysteine residues that become oxidized and thereby altering functions of the enzymes in a signaling pathway (Finkel 2012, Rhee et al. 2000, Tonks 2005). However, given the fact that increased levels of oxidative damages do accumulate during the aging process, one interesting new point of view proposes that intrinsic aging is caused by an inadequate response to endogenous ROS signals (Sohal and Orr 2012).
If ROS serve as signaling molecules as outlined above, it appears likely that ROS may also exert specific functions in promoting general health, and specifically lifespan. Since ROS at high doses unquestionably exert detrimental effects on cellular integrity, this insinuates that different levels of ROS, i.e. comparably low versus high amounts, may exert opposite effects on biological outcomes. In a more general sense, this kind of biphasic or non-linear response to potentially harmful substances was named “hormesis” (Southam and Ehrlich 1943). By today, the impact of hormetic effects on aging has been repeatedly proposed, with a range of stressors described (Calabrese and Baldwin 2002, Cypser and Johnson 2002, Rattan 2008, Mattson 2008, Lamming et al. 2004, Yanase et al. 1999). On a hypothetical basis the term was specified to mitochondrial hormesis or mitohormesis in 2006 (Tapia 2006), which after its experimental validation in parallel (Schulz et al. 2007) is repeatedly used in settings where mtROS act as sublethal stressors promoting lifespan, whereas higher doses increase lethality (Figure 1).
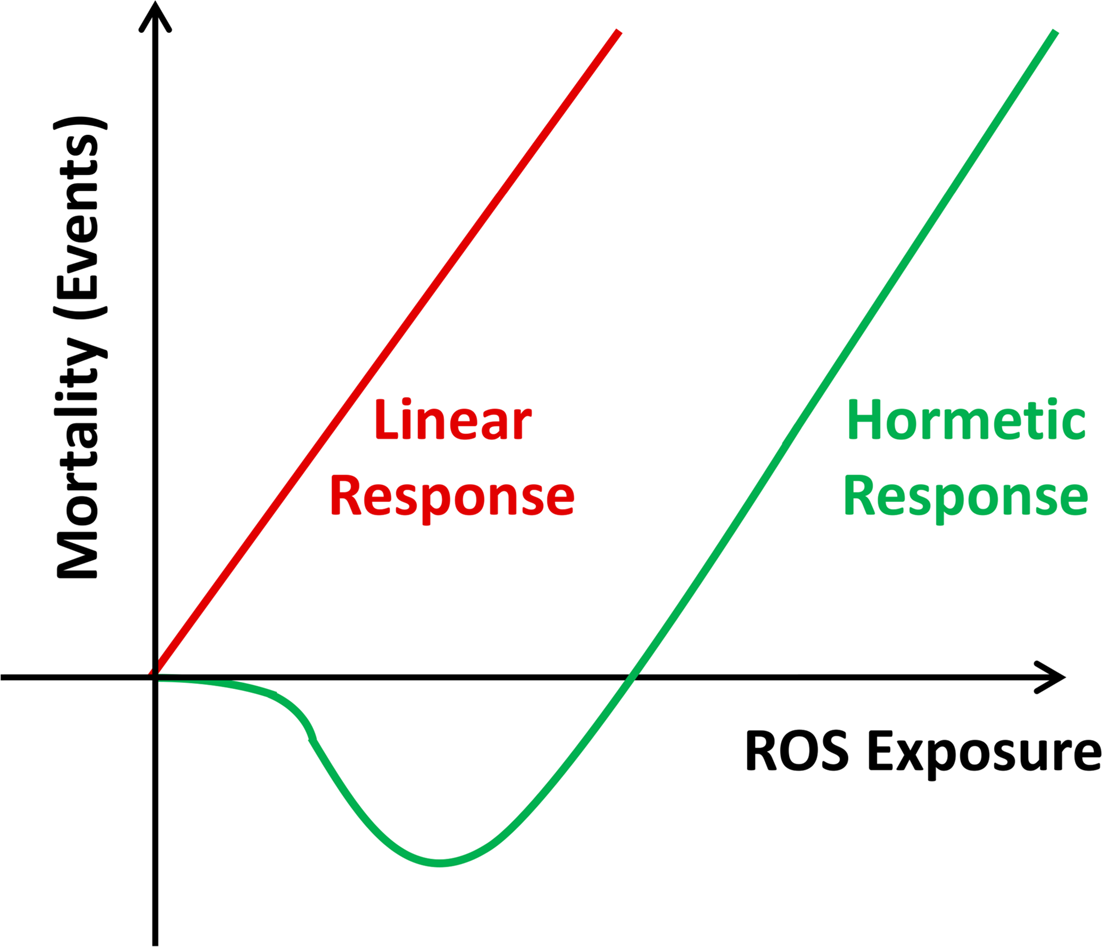
Mitochondrial Hormesis (Mitohormesis). While the Free Radical Theory of Aging suggests a linear dose-response relationship between increasing amounts of ROS and oxidative stress on the one hand, and mortality events on the other (red curve), the concept of mitohormesis indicates a non-linear dose-response relationship where low doses of ROS exposure decrease mortality, while higher doses promote mortality.
Aim of the review
We here aim to summarize the wide body of published evidence that refers to the biological relevance of mitohormesis mainly in regards to regulation of stress resistance and longevity, but also affiliated areas of interest. The majority of publications in this regard does not explicitly use the term mitohormesis but rather refers to dose-dependent or non-linear, mtROS-mediated signaling processes that hence reflect typical examples of mitohormesis.
2. CALORIE RESTRICTION (CR)
Calorie restriction (CR), being defined as a 10 − 50% reduction of ad libitum calorie uptake in the absence of malnutrition, is so far the most convincing intervention to delay both aging and the occurrence of age-related diseases in a variety of organisms, as reviewed elsewhere (Fontana et al. 2010). The first observation that laboratory rats maintained on dietary restriction not only showed an increased lifespan, but also seem to be healthier at higher age, dates back to 1935 (McCay et al. 1935). Since then, it has been frequently shown that CR is capable of extending median and maximum lifespan in various species from yeast to mammals (Lin et al. 2004, Lin et al. 2002, Schulz et al. 2007, Iwasaki et al. 1988), insinuating an evolutionarily conserved mechanism, as review elsewhere (Mair and Dillin 2008).
Nevertheless, it is still a matter of debate whether CR prolongs life expectancy in humans too as it is shown that people with average body mass tend to live longest (Berrington de Gonzalez et al. 2010), while CR in humans rather causes severe reduction of body mass (Holloszy and Fontana 2007). However, CR in humans clearly reduces diseases associated with aging, including cardiovascular diseases, cancer, and type 2 diabetes mellitus (DM type 2) (Takemori et al. 2011, He et al. 2012, Harvey et al. 2012, Willette et al. 2012, Ryan et al. 2012) as well as associated risk factors known to promote the before-mentioned diseases (Larson-Meyer et al. 2006, Heilbronn et al. 2006, Lefevre et al. 2009). One study found that CR reduces age-related mortality (which corresponds to only 54% of deaths) in rhesus monkeys, whereas no influence on overall mortality was reported (Colman et al. 2009). In contrast and unlike ad libitum fed control animals, monkeys on CR did not show any age-related impairment in glucose homeostasis, suggesting a reduction of prevalence of metabolic disorders like DM type 2. Another recent study on the same model organism found no changes in mortality following CR, whereas beneficial effects on health and morbidity were clearly observed (Mattison et al. 2012). It should be noted that the two studies have used diets that differed strikingly, also in regards to carbohydrate content. Due to the fact that both studies were not finished by the time this manuscript was prepared, future findings will have to show whether CR may affect overall mortality in these monkeys. Nevertheless, there is suggestive evidence that CR may also prolong life expectancy in primates and ultimately humans (Fontana et al. 2004, Heilbronn et al. 2006, Ingram et al. 2006, Weindruch 2006, Fontana and Klein 2007).
The concept of CR is based on an assumption postulated in the early 20th century, suggesting that there is an inverse correlation between the maximum lifespan of an organism and its metabolized nutritive energy (Rubner 1908). According to this, the Rate of Living hypothesis was formulated soon after by Raymond Pearl, insinuating that an increase in metabolic rate would decrease the lifespan of eukaryotes (Pearl 1928). A possible explanation for this was subsequently proposed within the FRTA by Harman, a hypothesis that became very popular and is frequently cited in aging research up to now (Harman 1956), as it is an explanation for CR which was hypothesized primarily to be a result of reduced oxidative stress and less oxidative cellular damage due to reduced metabolic rate (Sohal and Weindruch 1996).
However, more recent findings on the mechanistic basis of CR are in conflict with the FRTA. For instance, it is unclear whether CR actually does lead to a decrease in metabolic rate, i.e. oxygen consumption and/or heat production. A positive correlation for decreased metabolic rate and increase in longevity is found neither for metazoans like Drosophila and C. elegans, nor is it for mice (Hulbert et al. 2004, Lin et al. 2002, Masoro et al. 1982, Speakman et al. 2002). Rather, it has been reported that CR in C. elegans is associated with an increased metabolic rate (Walker et al. 2005, Schulz et al. 2007) as it is for Drosophila (Magwere et al. 2006, Piper et al. 2005b).
Since increased metabolic rates are necessarily linked to increased mitochondrial metabolism, it appears likely that these lifespan-extending processes may precipitate into increased production of ROS as an inevitable by-product of mitochondrial metabolism, as shown e.g. for glucose restriction (Schulz et al. 2007) and discussed in more detail below, reflecting a CR-associated prime example of mitohormesis.
3. MITOHORMETIC MECHANISMS OF ADAPTIVE RESPONSES
Notably, it was repeatedly reported that CR is capable of inducing stress defense mechanisms, particularly those which are involved in the detoxification of ROS, such as radical-scavenging enzymes and phase I and II biotransformation response enzymes, reflecting a number of putative mitohormetic responses (Koizumi et al. 1987, Semsei et al. 1989, Rao et al. 1990, Pieri et al. 1992, Youngman et al. 1992, Xia et al. 1995, Masoro 1998, Barros et al. 2004, Mahlke et al. 2011, Qiu et al. 2010, Rippe et al. 2010, Sreekumar et al. 2002, Park et al. 2012, Schulz et al. 2007, Zarse et al. 2012, Schmeisser et al. 2013b).
The independent observations of increased mtROS levels on the one hand and the induction of stress defense on the other, notably both in states of CR, raised the possibility that an initial induction of mtROS induce stress defense mechanisms culminating in secondarily decreased mtROS levels, as experimentally shown recently in a time-resolved manner (Zarse et al. 2012): In states of glucose restriction due to a constitutive genetic defect in the insulin/IGF-1 receptor DAF-2, a global decrease of mtROS levels in the steady-state was found. However, when analyzing an acute disruption of the same genetic pathway, a transient increase of mtROS was observed that secondarily induced defense mechanisms, ultimately reducing ROS levels in the steady state (Figure 2A). Blocking the initial ROS signal accordingly abrogated the induction of stress defense, as well as the steady-state reduction of ROS levels (Figure 2B). This indicates that the mitohormetic ROS signal is typically transient, and is reduced or even abolished in the steady-state due to an adaptive up-regulation of antioxidant enzymes and more globally stress defense. In other words, transiently increased ROS levels act to induce a vaccination-like response within the individual cell to lead to reduced ROS levels and better stress defense in the steady state (Figure 3).

Lifespan-promoting ROS signaling can occur transiently and hence requires time-resolved quantification. A) Disruption of the insulin/IGF-1 receptor, named DAF-2, in C. elegans extends lifespan. The constitutive daf-2 mutant exhibits reduced ROS levels. This has led to the conclusion that impairing DAF-2 primarily causes reduced ROS levels. However, as recently published (Zarse et al. 2012), the opposite is the case: When studying the acute effects of an RNAi-mediated daf-2 knockdown, a transient increase in ROS production was observed (“acute response”). As shown in the publication (Zarse et al. 2012), this ROS signal induces various endogenous ROS defense mechanisms that ultimately reduce ROS levels. This leads to a persistent reduction of ROS levels in daf-2 RNAi-treated worms in the steady state. This also exemplifies that quantifying ROS at an inappropriate time point may lead to opposing results: ROS determined during the acute response against RNAi would indicate increased levels, while ROS determined three days later during the steady-state would indicate reduced levels. B) Exogenously added antioxidants prevent the acute induction of a ROS signal (Zarse et al. 2012). The lack of this ROS signal leads to a complete lack of the original adaptive response shown in panel A. This causes higher steady-state ROS levels than in the absence of exogenous antioxidants which only can be explained in the framework of mitohormesis, while the linear dose-response would consider this phenomenon as paradoxical.
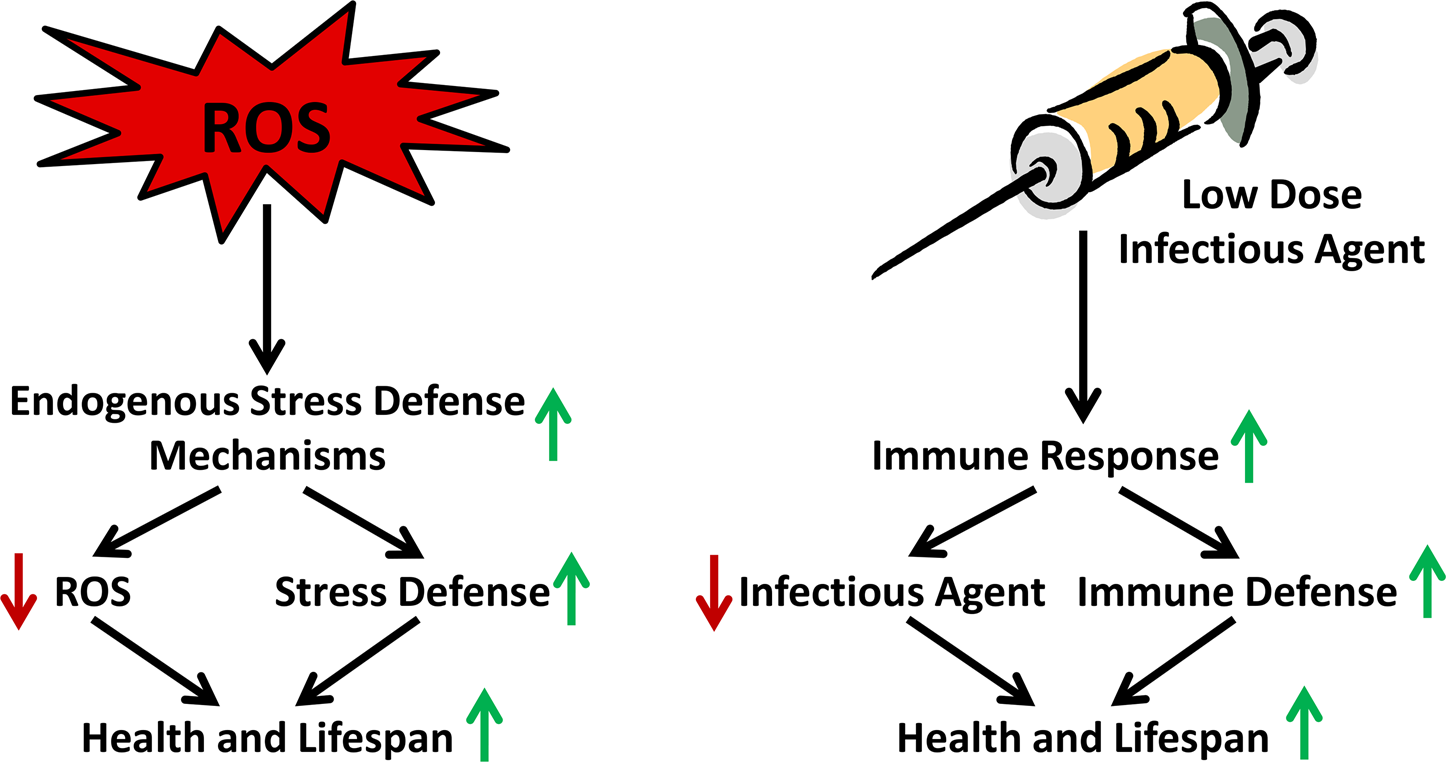
Transiently increased ROS levels cause a vaccination-like adaptive response that promotes endogenous ROS defense capacity. The figure exemplifies the organismal stress response to low-level and/or short-term ROS exposure in comparison to the long-standing vaccination process, where inactive or impaired microbes exert an organismal immune response leading to a long-lasting defense capacity against future infections.
This subsequent decrease in ROS due to an adaptive increase of detoxifying mechanisms has often been misinterpreted as being the primary result of CR, which as outlined above is not the case. Rather, a clear causal relationship between primarily enhanced ROS formation and activation of ROS defense mechanism under conditions of CR was described (Agarwal et al. 2005), which manifests the hypothesis that CR is an essential trigger of mitohormetic mechanisms as shown thereafter (Schulz et al. 2007). Moreover, carbonyl concentrations reflecting oxidative protein damage were found to be increased in the brains of mice shortly after initiation of CR, whereas steady-state concentrations were significantly lower than those of control group (Dubey et al. 1996). Furthermore, levels of F2-isoprostane, reflecting oxidized lipids, were found to be decreased in obese woman under modest calorie restriction after 5 days of intervention (Buchowski et al. 2012). According to this, adaptive response mechanisms seem to be likely the reason for the beneficial effects initiated by CR, which is also supported by more recent research (Schulz et al. 2007, Sharma et al. 2010, Zuin et al. 2010, Rattan and Demirovic 2010, Mesquita et al. 2010), also in a time-resolved manner (Zarse et al. 2012). In rodents that are exposed to CR for instance, an induction of antioxidant defense capacities has been frequently shown (Koizumi et al. 1987, Semsei et al. 1989, Rao et al. 1990, Pieri et al. 1992, Youngman et al. 1992, Xia et al. 1995, Masoro 1998, Barros et al. 2004, Mahlke et al. 2011, Qiu et al. 2010, Rippe et al. 2010, Sreekumar et al. 2002). Additionally, glucose restriction in yeast not only promotes lifespan but also decreases ROS levels although respiration was increased (Barros et al. 2004). In conflict with these findings, subsequent studies using the same models and interventions rather reported an increase in ROS production paralleled by enhanced respiration and elevated antioxidant enzyme activity (Schulz et al. 2007, Sharma et al. 2010, Zuin et al. 2010, Agarwal et al. 2005, Kharade et al. 2005, Piper et al. 2006). This suggests a relationship between increased respiration, ROS generation and the upregulation of ROS defense mechanisms, which in the end mediates longevity. Furthermore, lifespan-extending mutations in C. elegans are commonly associated with increased stress resistance and often also with increased metabolic activity (Lithgow et al. 1995, Vanfleteren and De Vreese 1995, Honda and Honda 1999, Murphy et al. 2003, Houthoofd et al. 2005, Dong et al. 2007).
As mentioned before, CR is able to delay the onset of a broad range of age-related diseases such as cancer, DM type 2, nephropathy, cataracts, hyperlipidemia, and hypertension (Fishbein 1991, Weindruch and Walford 1988). Therefore it seems possible, that the lifespan-extending effect of CR is linked to promotion of mean lifespan due to prevention of life-threatening disorders that reduce longevity. However, additional effects of CR on molecular processes improve cellular functions and therefore improve health-span per se have been observed, for instance the below mentioned activation of NF-E2-Related Factor 2 (NRF2) (Koizumi et al. 1987, Semsei et al. 1989, Rao et al. 1990, Pieri et al. 1992, Youngman et al. 1992, Xia et al. 1995, Masoro 1998, Barros et al. 2004, Mahlke et al. 2011, Qiu et al. 2010, Rippe et al. 2010, Sreekumar et al. 2002, Bishop and Guarente 2007). Another crucial role in CR and aging is attributed to the sirtuins, a conserved family of NAD+-dependent deacetylases as reviewed elsewhere (Baur and Sinclair 2006, Canto and Auwerx 2009) (see also chapter “Sirtuin signaling”).
An important factor regarding the effects of CR could also be thioredoxin, as it is shown to be essential for the lifespan extension in C. elegans under dietary deprivation and knockouts of eat-2, a genetic surrogate of nematodal CR (Fierro-Gonzalez et al. 2011). The oxidoreductase thioredoxin is not only involved in antioxidant response and redox regulation, but also acts as electron donor for metabolic enzymes and prevents aggregation of cytosolic proteins in the cell (Lillig and Holmgren 2007, Berndt et al. 2008). Thioredoxin gene expression is increased through NRF2 binding at the antioxidants responsive elements (AREs) and NRF2 is shown to be activated by ROS (Kim et al. 2001, Papaiahgari et al. 2006).
The activation of the transcription factor NRF2 from the leucine zipper family is indeed a crucial pathway to mediate mitohormesis. NRF2 binds to the DNA via AREs, which coordinate a stress response to ROS by boosting the expression of antioxidant proteins and phase I and II detoxification enzymes (Rushmore et al. 1991). Under unstressed conditions, NRF2 is sequestered in the cytoplasm by its specific repressor Kelch-like ECH-Associated Protein 1 (KEAP1), an actin-binding protein, which also targets NRF2 for proteasomal degradation (Itoh et al. 1999). KEAP-1 has redox-sensitive cysteine residues, with which it sensors oxidants and electrophiles, leading to abrogation of the NRF2/KEAP1 complex (Zhang 2006, Itoh et al. 2004). NRF2 then translocates into the nucleus, where it executes its transcriptional regulating functions (Jaiswal 2004) (Figure 4). While the putative functional orthologue of KEAP1 in C. elegans, XREB (Hasegawa and Miwa 2010), has not been examined further so far, the worm NRF2 orthologue SKN-1 similarly responds to oxidative stress by upregulating antioxidant and phase II genes which in the end promotes stress resistance and lifespan extension (An and Blackwell 2003, Bishop and Guarente 2007, Tullet et al. 2008), as it is shown for various other species (Sykiotis and Bohmann 2008, Motohashi and Yamamoto 2004, Leiser and Miller 2010, Lewis et al. 2010).

Overview on how ROS transcriptionally influence stress resistance and lifespan. High levels cause damage resulting in death of the cell and eventually the corresponding organism, whereas low levels are capable of activating transcription factors that mediate adaptive stress response culminating in increased lifespan.
Other transcription factors which are essential for lifespan extension due to various interventions are members of the Forkhead transcription factors (FOX) as well as heat shock factor 1 (HSF-1). FOXOs for example activate a number of target genes involved in cellular stress response and it has been shown, that mitohormetic upregulation of superoxide dismutase and catalase following oxidative stress is FOXO dependent (Kops et al. 2002, Nemoto and Finkel 2002, Brunet et al. 2004), whereas FOXAs are important mediators of development and facilitate the response to CR (Friedman and Kaestner 2006, Panowski et al. 2007). HSF-1 regulates the transcription of heat shock genes that encode proteins (HSPs) in response to heat and other stress, which is linked to protection against diseases and increased lifespan in model organisms (Akerfelt et al. 2010, Anckar and Sistonen 2011). HSPs are also linked to hormetic responses (Cypser and Johnson 2002). Other mechanisms specifically related to the stressors described in the chapters below will be mentioned at the appropriate position within this review (Figure 4).
4. REDUCTION OF SPECIFIC MACRONUTRIENTS
Macronutrients are carbohydrates, protein and fat (triacylglycerides), which consist of a few different monosaccharides (importantly including glucose), amino acids, and fatty acids, respectively. Metabolism of these macronutrients provides the majority of energy in form of ATP required by an organism. ATP generation out of fatty acids and most amino acids depends on mitochondrial OxPhos and therefore presence and consumption of oxygen. However, only glucose can be metabolized to generate ATP independently of the mitochondria and oxygen, hence without increasing ROS production. Nevertheless, the generation of ATP via OxPhos is considerably more efficient than anaerobic ATP production from glucose or specific amino acids: One mole of glucose metabolized via mitochondrial OxPhos provides 30 moles of ATP, compared to 4 moles of ATP due to exclusively glycolytic breakdown. This implies that restricting glucose may induce OxPhos and mitochondrial metabolism more efficiently than globally restricting calorie uptake.
On one hand, only a few studies have investigated whether it is possible to mimic the CR mediated effect on lifespan by reducing only selected macronutrients, so the evidence available is limited. On the other hand, some studies point out that it might be not the amount of calories affecting health span, but rather the reduction of specific nutrients, as reviewed elsewhere (Fanson et al. 2009, Mair et al. 2005, Piper et al. 2005a).
Studies of fat restriction in invertebrates are lacking, while a restriction of lipids without overall CR in mice does not affect their lifespan (Iwasaki et al. 1988). Feeding a low-carbohydrate/high-fat diet to mice reduced lifespan slightly, at least in comparison to a high-carbohydrate/low-fat diet (Keipert et al. 2011). However, it is likely that fat restriction has less potential to delay the onset of metabolic disorders than carbohydrate restriction (Ryan et al. 2007, Volek et al. 2009).
In mice, it has been shown that a reduced nutritional protein content extent lifespan (Stoltzner 1977, Leto et al. 1976, Fernandes et al. 1976) as it was shown for casein restriction in Drosophila (Min and Tatar 2006). Studies examining the restriction of the essential amino acid and glutathione precursor methionine found it not only to be lifespan extending. Increasing mitochondrial biogenesis and function, energy expenditure, stress resistance, aerobic capacity, insulin sensitivity, glutathione (GSH) and expression of glutathione-S-transferase (GST) as well as a decrease of oxidative stress and cell damage due to adaptive changes in methionine and GSH metabolism were also observed (Zimmerman et al. 2003, Miller et al. 2005, Perrone et al. 2010, Richie et al. 1994, Malloy et al. 2006, Sanz et al. 2006, Perrone et al. 2012, Tsai et al. 2010, Caro et al. 2008). Interestingly, co-treatment with an antioxidant, N-acetylcysteine, blocks some of the health-promoting effects of methionine restriction, accentuating a critical role for ROS with mitohormetic adaption processes in this regard (Elshorbagy et al. 2012, Sanchez-Roman et al. 2012).
As mentioned above, glucose (besides a few amino acids) is the only macronutrient that can be metabolized and generate ATP without producing ROS. In support of the mitohormesis concept, it is documented that glucose restriction initiates health-promoting and lifespan extending effects in rodents and various lower organisms, for instance in Drosophila (Mair et al. 2005) and yeast (Lin et al. 2002). In the latter, studies showed that lifespan extension depends on enhanced respiration and sirtuin activation, which is still a matter of heated debate (Lin et al. 2000, Kaeberlein et al. 2004, Agarwal et al. 2005, Guarente and Picard 2005, Smith et al. 2007, Roux et al. 2009). However, also sirtuin-independent pathways have been discussed (Barros et al. 2004, Roux et al. 2009).
To achieve depletion specifically of glucose metabolism in eukaryotic model organisms such as rodents or C. elegans and hence, to mimic a ketogenic diet (i.e. a very low carbohydrate content) and recapitulate metabolic hallmarks of CR in rodents, the glycolytic inhibitor 2-deoxy-glucose (DOG) is frequently used (Wick et al. 1957, Garriga-Canut et al. 2006, Lane et al. 1998, Ingram et al. 2004). DOG was found to be lifespan extending in C. elegans (Schulz et al. 2007), whereas and unexpectedly, increased mortality in rats was reported following chronic ingestion of DOG (Minor et al. 2010). It should be noted that shuttle mechanism for lactate and alanine may explain differential outcomes of glucose deprivation in metazoans and rodents, which however remains to be evaluated.
Similar as reported in yeast for media-based glucose restriction (Lin et al. 2002), DOG not only extends lifespan in C. elegans, but also increases respiration. However and unlike in S. cerevisiae, in C. elegans the effect seems to be independent of sirtuins. The authors suggested that the underlying mechanisms that lead to increased life expectancy are dependent on the AMP-dependent kinase (AMPK). AMPK acts as a highly conserved key regulator of energy metabolism within a cell, since functionally similar orthologues were found in lower species like flies and worms (Hardie et al. 2006, Apfeld et al. 2004, Greer et al. 2007a, Pan and Hardie 2002). AMPK is activated by metabolic stress like cellular lack of energy, resulting in upregulation of processes that produce energy, such as mitochondrial biogenesis. This leads to a compensation of the energy deficit and likely to additional health-promoting effects (Hardie et al. 2006).
Another approach to reduce glucose content within the cell is the impairment of GLUT-4 glucose transporters. Mice with disruption of GLUT-4 in both muscle and adipose tissue show fasting hyperglycemia, glucose intolerance, increased fatty acid turnover, and utilization. However, lifespan was not affected (Kotani et al. 2004). Strikingly, overexpression of GLUT-4 leading to an increase of cellular glucose does also not affect lifespan, whereas enhanced glucose abundance decreased lifespan significantly in C. elegans (McCarter et al. 2007, Lee et al. 2009b, Schlotterer et al. 2009, Schulz et al. 2007).
In humans, several approaches of varying macronutrients in the diet have been established, especially to lose weight in states of obesity. In this regard, low fat/high carbohydrate diets seem to be as efficient as low carbohydrate/high protein diets. Meta-analyses have shown that reducing carbohydrates may additionally reduce the risk of cardiovascular diseases (Nordmann et al. 2006, Hession et al. 2009, Volek et al. 2009, Wang et al. 2002). Furthermore, the reduction of several inflammation markers in overweight men and women with atherogenic dyslipidemia was reported (Forsythe et al. 2008). It was also shown that a ketogenic diet was capable to lower blood glucose levels in obese diabetic patients more effectively than overall CR (Hussain et al. 2012). In contrast, a Swedish study in over 43.000 middle-aged women found a significant increase in cardiovascular diseases following a ketogenic diet (Lagiou et al. 2012). The authors suggested that the protein source might play an important role and could contribute to this unexpected outcome. Interestingly, it was shown that a short-term low-carbohydrate/high fat intake may increase postprandial plasma glucose, suggesting a decrease in first-phase insulin secretion after the diet has started. However, other studies detect decreased glucose plasma levels on the long run, which could be due to adaptive mechanisms (Nobels et al. 1989, Boden et al. 2005).
5. CALORIE RESTRICTION MIMETICS (CRM)
CRMs are defined as pharmaceutical or naturally occurring compounds that may mimic the metabolic state of CR. Ideally these compounds would allow the organisms to eat normally, i.e. ad libitum, while the metabolic state would reflect reduced caloric uptake. The best studied compound in this regards, DOG has been introduced above. However, due to increased mortality in rats following chronic ingestion of DOG (Minor et al. 2010) this compound meanwhile appears of questionable usefulness. While beyond the immediate scope of this review, it should be noted that DOG as well as CR have been repeatedly shown to exert remarkable neuro-protective effects as reviewed elsewhere (Arumugam et al. 2006).
The phytochemical resveratrol is found to be CR mimetic as it potentially slows aging and certainly delays age-related diseases by activating sirtuins and also mitohormetic responses (Wood et al. 2004, Baur et al. 2006, Rubiolo et al. 2008) also based on a concept named xenohormesis (Howitz and Sinclair 2008). This is possibly linked to the fact that resveratrol may induce mtROS formation (Zini et al. 1999). Interestingly, genetic disruption of cellular mechanisms that target degradation of xenobiotics has been shown to result in lifespan extension in multiple species, suggesting adaptive response processes (Curran and Ruvkun 2007, Smith et al. 2008, Melo and Ruvkun 2012).
More specifically and focusing exclusively on mtROS formation, a recent study in C. elegans found that chemical inhibition of complex I of the mitochondrial electron transport chain (ETC) also mimics CR including increased physical activity and stress resistance, as well as extended lifespan. Interestingly, it was further shown that these complex I inhibitors extend lifespan independent of sirtuins and AMPK, but solely need a transient ROS increase to activate p38 MAP kinase and neuronal NRF2, suggesting that CR extends lifespan by inducing ROS formation (Schmeisser et al. 2013b). Consistently, C. elegans with genetic impairment of complex I, III, and IV are also long-lived (Dillin et al. 2002, Zuryn et al. 2010, Rea et al. 2007). In mice, inhibition of complex IV leads also to lifespan extension as well as elevated fat utilization, increased insulin sensitivity, and increased mitochondrial biogenesis (Deepa et al. 2012). Also, juglone, a known ROS generator and herbicide, has been reported to increase lifespan in low concentrations due to enhanced oxidative stress response (Heidler et al. 2010).
In this regard it is interesting to note that a significant number of pharmaceutically effective compounds, including phytochemicals like resveratrol (Zini et al. 1999), sulforaphane (Singh et al. 2005), niacin (Fukushima 2005), and berberine (Turner et al. 2008), as well as antidiabetic drugs like metformin (El-Mir et al. 2000) and PPARγ-activating thiazolidinediones (Brunmair et al. 2004b, Nadanaciva et al. 2007), and lastly cholesterol-lowering HMG-CoA-synthase inhibitors (“statins”) (Nadanaciva et al. 2007) and PPARα-activating fibrates (Brunmair et al. 2004a, Nadanaciva et al. 2007), have been found to inhibit mitochondrial complex I of the ETC. Since inhibition of complex I generates a mitochondrial ROS signal, these independent findings insinuate that this group of compounds exerts pleitropic effects, however sharing a common denominator by acting as potential CRMs which however needs further investigation.
6. IMPAIRED INSULIN/IGF-1 SIGNALING (IIS)
Mammalian insulin and IGF-1 (insulin-like growth factor 1) are peptide hormones produced in beta-cells of the pancreas and the liver, respectively. Insulin is a key regulator of glucose metabolism, especially involved in the regulation of cellular glucose uptake, fat metabolism, and food uptake. IGF-1 is produced following growth hormone (GH, a.k.a. somatotropin) mediated stimulation in the liver, and mediates childhood growth and anabolic effects in adults. Furthermore, most of the direct receptor-mediated effects of GH, i.e. IGF-1-independent effects, counteract insulin action. Insulin, IGF-1, and GH bind to different specific receptors to affect cellular functions. With a significantly lower affinity, insulin can activate the IFG-1 receptor, and vice versa.
Mice with impairment of GH or IGF-1 function and signaling show dwarfism and prolonged lifespan, somewhat resembling CR conditions (Quarrie and Riabowol 2004, Brown-Borg et al. 1996). Artificially increasing GH levels abrogates the lifespan extension and leads to elevated body size (Pendergrass et al. 1993, Steger et al. 1993). Furthermore, impairment of the neuronal IGF-1 receptor or heterozygous global disruption of this receptor increases murine lifespan and may be responsible for prevention of neurodegeneration and proteotoxicity (Holzenberger et al. 2003, Kappeler et al. 2008). Conversely, long-term IGF-1 exposure leads to decreased mitochrondrial function and cell viability in human fibroblasts (Bitto et al. 2010).
Impairment of the insulin receptor in humans is linked to insulin resistance, a state which is defined as an inadequate reduction of intracellular response to extracellular insulin stimulus (Kahn 1994). One main function of the insulin receptor activation due to extracellular insulin is the translocation of glucose transporter GLUT-4 and hence, glucose uptake in the cell. Accordingly, the outcome of insulin resistance is reduced glucose availability within the cell, which is associated with DM type 2 (Biddinger and Kahn 2006). This disease is linked to decreased lifespan also due to a number of secondary complications, including cardiovascular disease and an increased incidence of cancers (Kannel and McGee 1979, Franco et al. 2007, Coughlin et al. 2004).
Since in mice a global disruption of the insulin receptor knockout leads to embryonic lethality, muscle-specific knock-out mice were established to study the role of insulin receptor signaling. Noteworthy, muscle-tissue is the most relevant tissue in regards to glucose metabolism. Interestingly, those mice showed elevated fat mass, serum triacylglycerides and free fatty acids, but they neither experienced hyperglycemia nor the development of diabetes (Brüning et al. 1998). Moreover, they displayed enhanced glucose uptake in muscle cells in response to exercise like wild type mice do (Wojtaszewski et al. 1999). Lifespan data on those mice are not available, but mice with an adipose tissue-specific knockout have an increased mean and maximum lifespan (Blüher et al. 2003). Furthermore, mice with a global knockout in the insulin receptor substrate 1 (IRS-1) demonstrate increased resistance to several age-related pathologies and are long-lived, as are mice with neuronal knockout of IRS-2 and heterozygous global IRS-2 knockouts (Selman et al. 2008, Page et al. 2013, Taguchi et al. 2007). In addition, there are hints that specific mutations in the insulin receptor might also be associated with longevity in humans (van Heemst et al. 2005, Pawlikowska et al. 2009).
Other than mammals, invertebrates like C. elegans and Drosophila do not have distinct receptors for IGF-1 and insulin, but rather share a common receptor. Impaired insulin/IGF-1 signaling (but not its complete disruption) in invertebrates strikingly extends lifespan (Kimura et al. 1997, Clancy et al. 2001, Tatar et al. 2001).
C. elegans that carry a mutation within daf-2, the worm orthologue of the insulin/IGF-1 receptor, have a lifespan that is twice as long as in wild type worms (Kenyon et al. 1993). Recent studies have shown that the mitochondrial energy production is altered by impairment of DAF-2. Such mutants did not display the typical age-dependent decrease in mitochondria protein and bioenergetics competence, but a higher membrane potential and an increase in ROS, which is interestingly not associated with more, but less damage to mitochondrial DNA and protein (Brys et al. 2010, Zarse et al. 2012). The inhibition of IIS leads to lifespan extension due to changes in gene expression mediated by the FOXO transcription factor DAF-16 (Kenyon 2010). Reduction of IIS followed by lifespan extension and promotion of stress resistance in the worm not only requires DAF-16, it activates also SKN-1 in parallel, which facilitates the above mentioned beneficial adaptive response (Tullet et al. 2008); the same is true for HSF-1 (Kenyon 2005, Chiang et al. 2012). Also, mitochondrial L-proline catabolism plays an important role in that regard since it is upregulated by impaired DAF-2 signaling, which lead to a transient increase in ROS production mediating adaptive response processes to extend lifespan, proving the link between impaired IIS and mitohormesis (Zarse et al. 2012).
Assuming that impairment of the insulin/IGF-1 receptor reduces glucose uptake one could expect that the same lifespan extending mechanisms act here as they occur in regard to glucose restriction or overall CR (see above). In fact, there are several studies proposing shared processes and pathways involved in both interventions (Yechoor et al. 2004, Brooks et al. 2007, Katic et al. 2007, Russell and Kahn 2007, Westbrook et al. 2009, Zarse et al. 2012), while others propose independent mechanisms (Greer et al. 2007a, Lakowski and Hekimi 1998, Bartke et al. 2007, Houthoofd et al. 2003, Min et al. 2008, Bonkowski et al. 2009, Brown-Borg et al. 2002, Clancy et al. 2002).
7. AMP-DEPENDENT KINASE (AMPK) SIGNALING
AMPK acts as a sensor of available nutrients and hence energy that is regulated by the cellular AMP/ATP ratio and upstream kinases (Hardie et al. 2003). Whenever an energy deficit occurs and concurrently the AMP/ATP ratio rises, AMPK activates catabolic and represses anabolic processes. In other words, being activated by stress that inhibits ATP generation or increases ATP consumption, like glucose starvation or muscle contraction, AMPK inhibits energy consuming pathways and induces ATP-generating processes (Hardie et al. 2003, Salt et al. 1998, Winder and Hardie 1996).
AMPK exists as heterotrimeric complex consisting of a catalytic α-subunit and the regulatory β- and γ-subunits (Kemp et al. 2003). Activation of AMPK requires specific phosphorylation events by upstream kinases such as the serine/threonine protein kinase LKB1 within the catalytic domain of the α-subunit (Woods et al. 2003). Widely expressed, AMPK regulates food uptake in response to nutrient signals and hormones (Minokoshi et al. 2004) and is an important initiator of mitochondrial biogenesis (Zong et al. 2002, Winder et al. 2000), glucose and fatty acid uptake (Barnes et al. 2002, Habets et al. 2009), as well as β-oxidation (Merrill et al. 1997). In C. elegans, AMPK overexpression extends lifespan and is required for the lifespan extension due to impaired insulin/IGF-1 signaling (Apfeld et al. 2004). However, the role of AMPK in CR is less clearly established. The kinase appears to be necessary to mediate lifespan extension due to CR when food limitation starts in middle age employing a pathway that requires DAF-16/FOXO (Greer et al. 2007a). The direct activation of FOXO by AMPK has also been described in mammals (Greer et al. 2007b), linking it to oxidative stress response and therefore potentially mitohormesis.
Accordingly, AMPK is activated by DOG and involved in the induction of mitochondrial metabolism and hence the mitohormetic response (Schulz et al. 2007). Another example for an AMPK-activating substance is metformin, an antidiabetic drug and inhibitor of the mitochondrial complex I (El-Mir et al. 2000), which was found to be lifespan-extending due to AMPK-activation in C. elegans and mice (Onken and Driscoll 2010, Anisimov et al. 2008). Moreover, metformin was shown to promote adaptive processes, which are also involved in CR and oxidative stress response like activation of NRF2/SKN-1, culminating in increased life expectancy (Onken and Driscoll 2010). As described earlier, the CR mimetic resveratrol slows aging and delays age-related diseases by activating, besides sirtuins, also AMPK, again linking it to mitohormetic responses (Zini et al. 1999, Baur et al. 2006). It has been reported that many AMPK activators, including resveratrol and metformin, act by inhibiting mitochondrial function (Hawley et al. 2010). As a consequence of impaired mitochondrial function, the AMP/ATP ratio rises, leading to AMPK activation followed by increased mitochondrial biogenesis, respiration, β-oxidation, and finally increased ROS production (Schulz et al. 2007, Hardie 2011). It has moreover been proposed that ROS themselves are also capable of activating AMPK (Zmijewski et al. 2010, Alexander et al. 2010) and due to this, by acting up- and downstream of AMPK, the stress response may be further amplified. Interestingly, hypoxia has been shown to activate AMPK not by changing the AMP/ATP ratio, but rather by increased ROS production, since the activation is inhibited by antioxidants (Emerling et al. 2009).
However, recent findings suggest that mitochondrial ROS production may be more relevant than AMPK activation in regards to lifespan extension: consistent with the nuo-6 mutation in C. elegans (Yang and Hekimi 2010), inhibiting complex I of the respiratory chain by rotenone and other chemicals generates a ROS signal that extends lifespan in the absence of AMPK, sirtuins, or both (Schmeisser et al. 2013b). This indicates that ROS formation alone, i.e. in the absence of energy sensors, is still capable of promoting longevity. Consistently, it was shown that nematodes lacking AMPK live shorter and die prematurely in the dauer stage since their triglyceride stores are exhausted (Narbonne and Roy 2009, Xie and Roy 2012). The study of Xie and colleagues pointed out an important role for ROS in replacing essential AMPK functions: An increase in hydrogen peroxide activated the transcription factor hypoxia-inducible factor 1 (HIF-1; see also chapter “Hypoxia”), which is capable of stimulating key enzymes involved in the biosynthesis of fatty acids, leading to an increased survival of the dauer larvae (Xie and Roy 2012).
Hence, AMPK not only acts as regulator of metabolism, but also may play an important role in ROS signaling and adaptive response processes, which highlights the universal character of mitohormesis within cellular metabolism, whereas ROS signals still promote longevity even in the absence of AMPK.
8. TOR SIGNALING
The so-called “target of rapamycin” (TOR) pathway is known to be another major regulator of life expectancy by sensing nutrient and environmental signals (Pan et al. 2012). The mammalian TOR (mTOR) is a serine/threonine protein kinase and a member of the phosphatidylinositol 3-kinase-related kinase protein family which consists of two functionally distinct multi-protein complexes known as TOR complex 1 (TORC1) and TORC2 (Brunn et al. 1997). Each complex has an accessory protein; for TORC1 it is named regulatory-associated protein of mTOR (RAPTOR), for TORC2 rapamycin-insensitive companion of mTOR (RICTOR) (Hara et al. 2002, Laplante and Sabatini 2012). The major sensor of cellular inputs like nutrients, hormones, energy, and oxidative stress is TORC1, while TORC2 executes regulatory functions concerning cell survival and cytoskeletal polarity (Laplante and Sabatini 2012). TOR signaling has been shown to be regulated by AMPK, suggesting that both nutrient-sensing pathways are key regulators of mitochondrial metabolism (Gwinn et al. 2008).
Impairment of the TOR pathway is shown to be lifespan-extending in various organisms (Kaeberlein et al. 2005, Powers et al. 2006, Jia et al. 2004, Kapahi et al. 2004). The immunosuppressive and antifungal drug rapamycin acts as inhibitor of the TOR pathway as it inhibits TORC1 and is known to extend median and maximum lifespan of C. elegans and mice (Harrison et al. 2009, Robida-Stubbs et al. 2012). Notably, rapamycin not only extends lifespan, but also induces insulin resistance and impaired glucose metabolism (Lamming et al. 2012), again consistent with cellular energy deprivation and subsequent induction of mitohormesis. Similarly, mTOR signaling has been linked to oxidative nutrient metabolism in rodents (Sengupta et al. 2010).
Consistent with this, lifespan extension in yeast due to impaired TOR signaling is promoted by inducing expression of proteins from the respiratory complexes, mitochondrial ETC activity and overall mitochondrial metabolism (Pan and Shadel 2009, Powers et al. 2006, Bonawitz et al. 2007, Pan et al. 2011). In agreement with this, the influence of TOR on mitochondrial biogenesis and turnover is also found in mammals in a strongly tissue-dependent manner. In murine skeleton muscle tissue and cells for instance, rapamycin decreases expression of mitochondrial genes resulting in decreased oxygen consumption (Cunningham et al. 2007), whereas in fatty tissue opposite effects have been observed. Adipocyte-specific disruption of RAPTOR is linked to higher rates of mitochondrial uncoupling followed by enhanced energy expenditure, which protects the mice from gaining weight (Polak et al. 2008). Higher respiration rates and energy expenditure are also linked to increased expression of genes involved in OxPhos and beta-oxidation, especially in older mice (Katic et al. 2007).
There are several protein families like 4E-BP, ATG, and S6K, which modulate mitochondrial biogenesis downstream in the TOR signaling pathway. Consistently, the translational regulator 4E-BP is shown to have strong influence on the expression of OxPhos genes. In Drosophila, CR alters mRNA profiles via TORC1 in a 4E-BP dependent manner (Zid et al. 2009). Furthermore, TOR downstream acting proteins are not only involved in mitochondrial biogenesis, but also mitochondrial quality: ATG-5 mediates the degrading of dysfunctional mitochondria by autophagy (Twig et al. 2008), recently called “mitophagy” (Pua and He 2009). Notably, autophagy has also an important role in response to cellular stress, including starvation and pathogen infection, as well as in IIS mediated lifespan regulation (Kroemer et al. 2010, Levine et al. 2011, Toth et al. 2008, Hansen et al. 2008).
Due to the fact that TOR signaling plays a crucial role in mitochondrial biogenesis and turnover it is consequently involved in regulation of mtROS levels. For example, mouse mitochondria from skeletal muscle cells with impaired ATG-7 exhibit increased production of ROS (Wu et al. 2009b). This negative correlation between TOR signaling and ROS is also observed in yeast, where both tor-1 knock-out and rapamycin treatment caused increased levels of superoxide due to enhanced mitochondrial biogenesis, strikingly coupled with less oxidative damage within the cell (Pan et al. 2011). This research manifests that ROS stimulation due to TOR acts also as mitohormetic stimulus and that mitohormesis is also in this regard the key mediator of lifespan extension (Pan 2011). Impaired TOR signaling through either genetically inhibited TORC1 or the usage of rapamycin is also known to activate SKN-1 and DAF-16 in C. elegans, mediating increased stress resistance and longevity (Robida-Stubbs et al. 2012).
It is assumed that TOR is another key mediator of CR since it has been shown that yeast and Drosophila carrying a deletion in the TOR gene do not benefit of CR in regards to lifespan extension (Kaeberlein et al. 2005, Kapahi et al. 2004). To evidence this hypothesis and point out whether the TOR pathway is involved in CR benefits, further research is strongly needed.
9. SIRTUIN SIGNALING
Sirtuins are NAD+-dependent deacetylases that catalyze the removal of acetyl groups from lysine residues of specifically histones and other proteins. They modulate cell-protective mechanisms such as oxidative stress defense, DNA repair, protein folding, energy utilization, and autophagy (Haigis and Sinclair 2010). The first identified member of this protein family was named silent information regulator 2 (SIR2) (Sinclair et al. 1997, Kaeberlein et al. 1999), giving rise to the term “sirtuins”. By today, seven mammalian orthologues have been found, named SIRT1 to SIRT7 (Blander and Guarente 2004), whereas SIRT1 and SIRT3 are the closest orthologues to SIR2 (Merksamer et al. 2013). Sirtuins are linked to longevity, since overexpression has been shown to extend lifespan in yeast (Kaeberlein et al. 1999) as well as in worms (Tissenbaum and Guarente 2001, Viswanathan and Guarente 2011, Mouchiroud et al. 2013, Ludewig et al. 2013, Schmeisser et al. 2013a) and flies (Rogina and Helfand 2004, Bauer et al. 2009). However, others could not confirm the results in C. elegans and Drosophila (Burnett et al. 2011), whereas one study found sirtuin overexpression only in the fat body of relevance for longevity (Banerjee et al. 2012). Sirtuins were found to be necessary to mediate lifespan extension due to CR (Lin et al. 2000, Guarente and Picard 2005, Boily et al. 2008), whereas others found no such connection (Kaeberlein et al. 2004, Smith et al. 2007, Schulz et al. 2007). In addition, a recent publication pointed out an important role for p53 modulating SIRT1 during CR, as reviewed elsewhere (Tucci 2012).
However, the role of sirtuins in oxidative stress and mitohormetic responses has been implicitly discussed also in this regard (Lin et al. 2000), supported by the observations, that sir2 overexpression rescues the short lifespan phenotype due to hydrogen peroxide treatment in yeast (Oberdoerffer et al. 2008) and that SIRT3 is necessary to mitigate oxidative stress during CR (Someya et al. 2010, Qiu et al. 2010). There is evidence that the mammalian SIRT1 is involved in mediating oxidative stress response, as it directly deacetylates several FOX members (Brunet et al. 2004, Motta et al. 2004, van der Horst et al. 2004). In contracting muscle cells SIRT1 mediates the protection against oxidative stress via enhanced expression of SOD-2 (Pardo et al. 2011). Correspondingly, SIRT2 activates FOXO3a, which promotes resistance to hydrogen peroxide (Wang et al. 2007). Moreover, SIRT1 has been also shown to activate peroxisome-proliferator-activated receptor (PPAR) gamma co-activator-1 alpha (PGC-1α) (Rodgers et al. 2005), a transcriptional co-activator that promotes mitochondrial biogenesis and expression of antioxidant genes including calalase, SOD, and glutathione peroxidase (St. Pierre et al. 2006). Furthermore, SIRT1 suppresses the inducible nitric oxide synthase (iNOS) and thus may decrease cellular ROS levels (Lee et al. 2009a). The catalytic activity of SOD-2 is dependent of the mitochondrial SIRT3, which is also capable of enhancing SOD-2 activity (Qiu et al. 2010). Accordingly, genetic impairment of SIRT3 in mice leads to higher ROS levels, genomic instability, and susceptibility to cancer (Kim et al. 2010), establishing SIRT3 as anticarcinogenic protein by improving stress response, which is supported also by more recent research (Bell et al. 2011, Finley et al. 2011). Consistently, SIRT6 has been shown to promote DNA repair in response to oxidative stress (Mao et al. 2011). Genetic impairment of SIRT6 results in genetic instability and premature aging (Mostoslavsky et al. 2006), whereas overexpression promotes lifespan extension, at least in male mice (Kanfi et al. 2012). Finally, SIRT7 mediates oxidative stress response as well, since cardiomyocytes of SIRT7 knockout mice are more sensitive to hydrogen peroxide treatment (Vakhrusheva et al. 2008, Calabrese et al. 2007).
From a traditional viewpoint, these findings support the notion that sirtuins promote health and lifespan, at least in parts, via increased resistance towards ROS (Webster et al. 2012) and particularly mitohormetic response processes (Merksamer et al. 2013). As outlined in more detail elsewhere (Merksamer et al. 2013), physiological stressors including CR may decrease the activity of antioxidant enzymes like SOD by acetylation processes resulting in hyperacetylation (Hirschey et al. 2010, Hirschey et al. 2011a, Hirschey et al. 2011b, Ozden et al. 2011). The following increase in ROS would activate defense mechanisms against oxidative stress, resulting in lower ROS levels in the steady state. Notably, such stresses have been also shown to increase SIRT3 expression, suggesting subsequently increased deacetylation of SOD and other mitochondrial proteins to counteract chronically increased ROS generation (Hirschey et al. 2010, Merksamer et al. 2013).
Very recently, an alternate mechanism linking sirtuin signaling to ROS-mediated lifespan extension has emerged (Schmeisser et al. 2013a): Sirtuins require NAD+ as a cofactor, and accordingly produce nicotinamide. This product becomes methylated to 1-methylnicotinamide, which itself serves as a substrate for an aldehyde oxidase to produce hydrogen peroxide. The latter acts as a ROS signal to execute sirtuin effects, since disruption of either the methylase or the oxidase fully prevents sirtuin-mediated lifespan extension (Schmeisser et al. 2013a), also implying that sirtuin-mediated deacetylation processes may be of limited relevance regarding lifespan regulation.
10. HYPOXIA
Hypoxia is an environmental state that is characterized by decreased environmental availability of oxygen, which typically leads to reduced mitochondrial respiration rates and a variety of changes on the molecular level (Semenza 2012), notably including increased mtROS production (Kulisz et al. 2002). The master regulator of hypoxia-mediated transcriptional changes is HIF-1, a highly conserved transcription factor that promotes survival under hypoxic stress (Shen and Powell-Coffman 2003, Semenza 2012). Under normal oxygen conditions, the mammalian HIF-1α subunit is hydroxylated and targeted for proteasomal degradation by the von Hippel-Lindau tumor suppressor protein (VHL) (Kim and Kaelin 2003). This signaling pathway seems to be highly conserved, since C. elegans hif-1 and vhl-1 genes encode homologs of HIF-1α subunit and VHL (Shen et al. 2005). However, it was shown that low oxygen atmosphere and decreased respiration is capable of increasing lifespan of C. elegans (Adachi et al. 1998), probably via stabilization of HIF-1 (Lee et al. 2010, Mehta et al. 2009, Zhang et al. 2009). This activation of the hypoxic signaling pathway was found to promote lifespan independently of CR or impaired IIS (Kaeberlein and Kapahi 2009, Mehta et al. 2009) as it is shown that CR due to deprivation of bacteria and genetically induced through mutation in eat-2 increases lifespan in hif-1 knockout nematodes, and impairment of DAF-2 is also able to promote longevity in those animals (Mehta et al. 2009). Notably, it was reported that loss of HIF-1 causes longevity as well (Zhang et al. 2009, Chen et al. 2009). In one study, DAF-16 seems to be essential for lifespan extension, indicating a mechanism similar to reduced IIS (Zhang et al. 2009), whereas others found no such connection, possibly insinuating that HIF-1 acts as a negative regulator of longevity in a pathway upstream of the endoplasmic reticulum (ER) stress response and downstream of CR and TOR signaling (Chen et al. 2009).
The unquestionable influences of HIF-1 on aging have initiated several competing hypotheses: One explanation could be that HIF-1 down-regulates mitochondrial activity (Papandreou et al. 2006, Semenza 2011), as this is shown to be lifespan extending through RNAi-mediated knockdown of several mitochondrial proteins (Tormos and Chandel 2010, Rea et al. 2007, Dillin et al. 2002). Alternatively, HIF-1 could act in regards to stress response, like NRF2 or FOXO, especially since it is shown that HIF-1 and DAF-16 share various target genes (McElwee et al. 2004). In mammals, there are also links to TOR signaling and ER unfolded protein response (UPR) with mTOR signaling being reduced by hypoxia and HIF-1 translation being dependent on TOR (Stein et al. 1998, Wouters and Koritzinsky 2008), whereas both hypoxia and TOR are known to activate UPR (Romero-Ramirez et al. 2004).
Interestingly, mitochondrial-derived ROS during hypoxia lead to HIF-1 stabilization in cultured cells (Chandel et al. 1998), as well as activation of c-Jun N-terminal kinase 1 (JNK1), p53, and NF-κB (Chandel et al. 2000a, Chandel et al. 2000b). Moreover, in C. elegans, knockdown of genes encoding respiratory chain components and mutations in such, like clk-1 and isp-1, lead not only to decreased respiration rates, but also to a mild increase in ROS formation, which is responsible for HIF-1 stabilization and longevity of the nematodes (Lee et al. 2010, Yang et al. 2009). Increased ROS levels under hypoxic conditions in C. elegans (Miller et al. 2011, Miller and Roth 2007) as well as in cultured cells (Guzy and Schumacker 2006) occur in a HIF-1 dependent manner. A recent study in C. elegans found also an important role of DAF-16 in this regard, since it is delocalized to the nucleus and necessary to extend lifespan under hypoxic conditions (Leiser et al. 2013). However, in this study, lifespan extension did not require SIR-2.1, AAK-2, SKN-1, or CEP-1, the worms’ orthologues of sirtuins, AMPK, NRF2 and p53, respectively. On the other hand, roles for the sirtuins in HIF-1 deacetylation, AAK-2 in adaption to anoxia, and CEP-1 acting downstream of HIF-1 have been described earlier (Dioum et al. 2009, Zhong et al. 2010, Lim et al. 2010, Leiser and Kaeberlein 2010, Larue and Padilla 2011, Sendoel et al. 2010). As mentioned above, a mild increase in oxidative stress leads to stabilization of HIF-1 followed by increased survival of C. elegans AAK-2/AMPK mutants due to HIF-1-dependent activation of genes involved in fatty acid biosynthesis (Xie and Roy 2012). This metabolic adjustment pointed out an important role for HIF-1 and ROS in compensating AMPK functions.
Nevertheless, studies that investigated the influence of hypoxia on mammalian aging are rare. This is not only because the mechanisms according to hypoxia in mammals are much more complex than in lower organisms like C. elegans, but also due to HIF-1α is involved in tumor growth and cancer development, notably also by altering glucose metabolism (Semenza 2012, Semenza et al. 1994). Discovered by its ability to increase erythropoetin production, HIF-1α was associated with the VHL hereditary cancer syndrome, a heterozygous disease characterized by development of various malign tumors in the kidneys, retina, and the central nervous system with an increase in HIF-1α being a negative predictor in metastatic tumors (Wang et al. 1995, Kaelin 2002, Semenza 2010).
However and to our best knowledge, the first evidence that links HIF-1 to longevity and aging per se only dates back to 2009 (Mehta et al. 2009), so future research will properly establish the role of oxygen availability in the aging process and bring hypoxic mechanisms and connections to other pathways to light.
11. TEMPERATURE STRESS
As early as in 1908 it was hypothesized that body temperature may be linked or even determine life expectancy (Loeb 1908). A few years later, the hypothesis was experimentally supported by showing that lowering temperature extends lifespan of poikilothermic Drosophila (Loeb and Northrop 1916). Subsequently, benefits from exposure to lowered temperature regarding lifespan have been shown in other organisms like C. elegans or fish (Klass 1977, Liu and Walford 1966) and notably also in homoeothermic (warm-blooded) animals like rats and mice (Holloszy and Smith 1986, Conti et al. 2006). On the other hand, increasing ambient temperature or mild heat stress are also linked to increased lifespan in various organisms (Shama et al. 1998, Wu et al. 2009a). As mentioned above, HSPs are major regulators of response to heat stress in almost all organisms investigated ranging from bacteria to mammals (Lindquist and Craig 1988, Fargnoli et al. 1990, Udelsman et al. 1993, Lithgow et al. 1995, Rea et al. 2005). HSPs consist of a large number of proteins, often being classified according to their molecular weight: HSP40, HSP60, HSP70, HSP90, HSP110 (with 40, 60, 70, 90, and 110 kilo-daltons in size, respectively) and the small HSPs represent the majority of HSPs (Li and Srivastava 2004). Some HSPs are also known as chaperones, playing crucial roles in the UPR to prevent polypeptides from aggregating into non-functional structures (Calderwood et al. 2009, Jazwinski 2005, Parikh et al. 1987), which has also been reported to play a role in lifespan regulation (Calfon et al. 2002, Henis-Korenblit et al. 2010, Yoneda et al. 2004). Transcriptionally regulated by HSF-1, HSPs have been unquestionably linked to hormetic processes (Akerfelt et al. 2010, Cypser and Johnson 2002). For instance, increased expression of HSP-70 family members following activation of HSF-1 due to a variety of stressors leads to protection against the latter, notably including ROS (Westerheide and Morimoto 2005, Raynes et al. 2012). Conversely, HSF-1 depletion shortens lifespan in C. elegans, as overexpression increases longevity and is required for the lifespan extension due to impaired insulin signaling (Hsu et al. 2003). The same study found that DAF-16 is necessary for extending lifespan in hsf-1 overexpressing worms, suggesting that both transcription factors might synergistically act to exert their beneficial effects (Hsu et al. 2003) (Figure 4). Hormetic heat stress is linked to improved mitochondrial function (Shama et al. 1998), which is required for ROS defense (Grant et al. 1997). Notably, long-lived C. elegans daf-2 mutants are resistant to thermal and oxidative stress and display increased expression of various HSPs and antioxidant and drug-metabolizing enzymes (McElwee et al. 2007, Lithgow and Walker 2002).
A mechanism for the observed increased lifespan at low temperatures was recently suggested in a study using C. elegans, pointing out an important role for a member of the transient receptor potential (TRP) family of cationic channels, TRPA-1 (Xiao et al. 2013). TRPA-1 alters its permeability to Ca2+, Na+, and K+ when activated by temperatures around 17°C or lower (Clapham 2003, Story et al. 2003). Worms that lack TRPA-1 have a shorter lifespan when exposed to cold in comparison to wild type animals, whereas overexpression of trpa-1 leads to increased lifespan at 15°C and 20°C, but not under warm (25°C) conditions. These effects were dependent on a calcium influx, which activates the calcium-sensitive protein kinase C (PKC) and the serine/threonine-protein kinase 1 (SGK-1). Interestingly, DAF-16/FOXO has been shown to be necessary to promote longevity in this regard. The TRPA-1 pathway induces nuclear activity of DAF-16, surprisingly without stimulating its nuclear translocation. The well-known fact that calcium influx increases ROS generation in mitochondria, as reviewed elsewhere (Brookes et al. 2004, Csordas and Hajnoczky 2009), insinuates that these ROS act as signal molecules to activate DAF-16 and promote longevity in this regard. Another study reported that hypothermia causes not only calcium influx into mitochondria, but also leads to a redox imbalance caused by an increase in ROS concentration (Brinkkoetter et al. 2008). Thus, mitohormetic processes could be also responsible for the lifespan extension following exposure to cold temperatures.
12. PHYSICAL ACTIVITY
Physical inactivity promotes the onset of a variety of diseases like obesity, cardiovascular disease, DM type 2, and cancer. Consistently, regular physical activity unquestionably exerts beneficial or preventive effects on the above mentioned diseases, and additionally delays depressive symptoms, neurodegeneration (including Alzheimer's disease), and general aging (Warburton et al. 2006, James et al. 1984, Hu et al. 2001, Brown et al. 2012, Lanza et al. 2008, Manini et al. 2006, Powers et al. 2011). Exercise is not only linked to enhanced mitochondrial biogenesis and oxidative metabolism, but also to increased generation of mtROS (Powers and Jackson 2008, Chevion et al. 2003, Davies et al. 1982, Alessio and Goldfarb 1988, Alessio et al. 1988). Thus, and because of its obvious beneficial effects in regards to health and aging, make it a paradigm of adaptive response processes and finally mitohormesis (Radak et al. 2008, Radak et al. 2005, Ji et al. 2006, Watson 2013). However, similar to physical inactivity, overtraining or excessive exercise represents the other end of the hormesis curve as the adaption process is inhibited, leading to incomplete recovery (Chevion et al. 2003) and resulting in maladaptation and possibly increased risk of diseases (Alessio et al. 1988).
To our knowledge, the first direct evidence that increased ROS production following exercise may act as stimulus to activate mitochondria biogenesis and mediates potential health-beneficial effects dates back to 1982 (Davies et al. 1982). An indirect clue was already given in 1971 with an antioxidant, namely vitamin E, causing unfavorable effects on the endurance performance of swimmers (Sharman et al. 1971). Since then, a bulk of studies (in most cases inadvertently) proved the hypothesis that ROS are required for the health-promoting effects of physical activity, causing an increase in antioxidant defense mechanisms and with this, prolong health span and mean lifespan (Crawford and Davies 1994, Davies 1986, Kim et al. 1996, Marzatico et al. 1997, Balakrishnan and Anuradha 1998, Ji et al. 2006, Powers and Lennon 1999, Niess et al. 1999, Hollander et al. 2001, Higuchi et al. 1985, Gomez-Cabrera et al. 2008b, Quintanilha 1984, Vincent et al. 1999, Boveris and Navarro 2008).
One of the main changes due to regular physical activity is the increase in mitochondria energy metabolism. Exercise activates PGC-1α, which is capable of controlling mitochondrial gene expression via NRF1 and the mitochondrial transcription factor A (TFAM). This mediates enhanced replication of mitochondrial DNA, leading to increased mitochondrial biogenesis and efficient muscle contraction (Nikolaidis and Jamurtas 2009, Akimoto et al. 2005, Baar 2004, Arbogast and Reid 2004). Furthermore, PGC-1 promotes the response to oxidative stress through activation of NRF2 and induction of antioxidant enzyme expression (St. Pierre et al. 2006). Another important point is the massive consumption of ATP followed by an increase in AMP, which activates AMPK, leading again to induction of PGC-1 and enhanced mitochondrial biogenesis (Bergeron et al. 2001, Atherton et al. 2005). This increase in mitochondrial metabolism leads to enhanced oxygen consumption in muscle fibers followed by lower intracellular oxygen tension during exercise, promoting ROS generation (Franco et al. 1999, Puntschart et al. 1996). There are also other so-called contraction-induced changes that stimulate ROS production in muscle, for instance increased CO2 tension, decreased cellular pH, and rise in muscle temperature (Arbogast and Reid 2004). The main source of ROS during exercise is probably skeletal muscle (Davies et al. 1982, Powers and Jackson 2008), but other tissues such as heart, lungs, and blood are also likely to be important contributors (Powers and Jackson 2008, Nikolaidis and Jamurtas 2009). On cellular level, mtROS were considered to be the predominant fraction of ROS produced during physical activity over decades (Koren et al. 1983, Davies et al. 1982), whereas recent research pointed out also important roles for nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, phospholipase A2, and xanthine oxidase (Powers et al. 2011).
ROS signals caused by a single bout of exercise only already activate antioxidant defense enzymes like mitochondrial SOD and inducible nitric oxide synthase (iNOS) (Hemmrich et al. 2003, Hollander et al. 2001). Regular exercise leads to proper adaptation to oxidative stress due to upregulation of diverse SODs, catalase, HSPs, and glutathione peroxidase (Powers and Lennon 1999, Leeuwenburgh and Heinecke 2001, Franco et al. 1999, Puntschart et al. 1996). The second line of antioxidant response which includes repair systems is important to minimize the damaging effects of ROS and is also activated through regular physical activity (Crawford and Davies 1994, Davies 1986), assigning important roles for proteasomal degradation and DNA repair enzymes (Radak et al. 2000, Radak et al. 1999, Radak et al. 2003).
Correspondingly, there is convincing evidence that supplementation of antioxidants is useless (Gey et al. 1970, Keren and Epstein 1980, Maughan 1999, Theodorou et al. 2011, Yfanti et al. 2010) or even harmful for athletes, potentially abolishing the beneficial effects on endurance performance, immune status, muscle development, and prevention of diseases (Gomez-Cabrera et al. 2008a, Strobel et al. 2011, Ristow et al. 2009, Marshall et al. 2002, Khassaf et al. 2003). For instance, athletes supplementing vitamin C and E did not display an induction of insulin sensitivity and endogenous antioxidant defense regulators due to exercise as seen in the control group (Ristow et al. 2009). It was shown that enhanced mitochondrial biogenesis and with this, increased respiration and ROS generation according to physical activity is prevented by co-treatment with antioxidants, leading to the inhibition of the beneficial mitohormetic response (Gomez-Cabrera et al. 2008a, Strobel et al. 2011, Kang et al. 2009, Fischer et al. 2006, Ristow et al. 2009). Furthermore, studies proved the harmful effect of antioxidants in regards to performance as it has shown to delay the recovery process (Close et al. 2006, Jackson 2008). Hence, supplementation of antioxidants should not be recommended to healthy athletes due to evidence that antioxidants have counter-productive effects on performance, health, and the onset of diseases.
13. OUTLOOK
All the above mentioned interventions are able to promote health-and lifespan in a variety of model organisms via mitohormetic processes (Figure 5). Future research will have to show whether these interventions will be capable of slowing aging and prolonging health span also in humans, in case it is not been shown yet. However, it seems unquestionable that the hypothesis of mitohormesis is, at least in parts, suitable to explain how the aging process could be beneficially influenced. Of course, mitohormesis cannot be considered in isolation to understand aging, which is to describe unfortunately beyond the topic and space limitations of this review. Notably, recent evidence suggests that stem cell aging is linked to impaired ROS signaling, i.e. that low levels of ROS production may prevent stem cell decline (Owusu-Ansah and Banerjee 2009, Owusu-Ansah et al. 2008, Morimoto et al. 2013). Given the eminent role of stem cell maintenance in the prevention of aging, it will be interesting to see whether the emerging link to increased ROS levels can be expanded. Secondly, ROS-mediated nitric oxide-signaling appears to be an increasingly expanding field of mitochondrial biology and disease control (D'Antona et al. 2010, Nisoli et al. 2005). Moreover, processes like proteostasis and mitochondrial UPR exert, while beyond the scope of this review, significant links to ROS-dependent signaling events, suggesting an overarching ROS-triggered mechanism in cellular and systemic quality control (Taylor and Dillin 2013, Balch et al. 2008). Furthermore, it should be emphasized that ROS signaling is a rather established mechanism in plant biology research (Mittler et al. 2011), which could not possibly be covered in the current review.

A non-exhaustive overview on lifespan-extending interventions linked to mitohormetic ROS signaling. As outlined in the text of the current review, a number of apparently diverse interventions lead to a mitohormetic response mechanism, insinuating that distinct molecular pathways culminate in a common mechanistic denominator by promoting a ROS-dependent stress response.
Related to the theory of mitohormesis is the Epigenetic oxidative redox shift (EORS) theory of aging, proposing a metabolic shift away from the use of mitochondrial energy towards reliance on glycolysis as a cause of aging. This is due to epigenetic mediators influencing histone deacetylases as well as histone acetylases and DNA methyltransferases (Brewer 2010, Ghosh et al. 2012). The shift in the oxidized direction of relevant oxidants and reductants, such as cysteine/cystine or GSH/GSSG occurs with aging and is initiated by low demands of mitochondrial produced energy. The low energy demand is caused by low physical or mental activity, initiating a vicious cycle of oxidized signaling molecules, transcription factors, membrane receptors, and epigenetic transcriptional regulators. This results in the inability to respond to energy demands and stress, leading to typical age accompaniments like cell death and organ failure. Notably, this EORS occurs upstream of the commonly observed increase in ROS damage to macromolecules (Brewer 2010, Ghosh et al. 2012).
Another interesting approach which further extends the mitohormesis concept is the Redox stress hypothesis of aging (Sohal and Orr 2012), according to which ROS have essential functions in regulating protein activity. The theory distinguishes between young and old organisms, proposing that in the first part of life thiol redox potential is high, whereas ROS generation is relatively low. In older organisms, ROS formation increases, which would lead to a pro-oxidized shift in redox state and overoxidation of thiols, resulting in loss of sensitivity and coordination among the regulatory processes, a progressive decline of function and finally death. Notably and like mitohormesis, the hypothesis relegates cumulative damages through ROS and antioxidant defense to an auxiliary status, with little impact on altering redox-sensitive signaling (Sohal and Orr 2012).
Taken together, mitohormesis unifies a significant number of lifespan-regulating molecular pathways, and may, dependent on additional scientific evidence, become a common denominator in aging research.
Footnotes
ACKNOWLEDGMENTS
The authors apologize to all colleagues whose work could not be cited solely due to the lack of space. We thank many members of the Ristow lab for interesting discussions regarding the topics outlined in this review. Funding sources: German Research Association (DFG) Graduate School of Adaptive Stress Response (#1715); Jena Centre for Systems Biology of Ageing (JenAge) funded by the German Ministry for Education and Research (Bundesministerium für Bildung und Forschung; support code BMBF 0315581); European Foundation for the Study of Diabetes (EFSD).