
Abstract
Risk prediction and dose limits for human radiation exposure are based on the assumption that risk is proportional to total dose. However, there is concern about the appropriateness of those limits for people who may be genetically cancer prone. The TP53 gene product functions in regulatory pathways for DNA repair, cell cycle checkpoints and apoptosis, processes critical in determining ionizing radiation risk for both carcinogenesis and teratogenesis. Mice that are deficient in TP53 function are cancer prone. This review examines the influence of variations in TP53 gene activity on cancer and teratogenic risk in mice exposed to radiation in vivo, and compares those observations to the assumptions and predictions of radiation risk inherent in the existing system of radiation protection. Current assumptions concerning a linear response with dose, dose additivity, lack of thresholds and dose rate reduction factors all appear incorrect at low doses. TP53 functional variations can further modify radiation risk from either high or low doses, or risk from radiation exposures combined with other stresses, and those modifications can result in both quantitative and qualitative changes in risk.
THE LNT HYPOTHESIS AND RISK PREDICTION
The linear no-threshold (LNT) hypothesis is the fundamental basis for the prediction of risk from radiation exposure, and forms the basis for radiation protection practices (ICRP 1991). Dose limits for human exposure reflect this assumption that risk is proportional to total dose, without a threshold. However, radiation protection practices also utilize a number of additional concepts, derived from or auxiliary to the hypothesis, to predict the risk of radiation exposure. The most basic concept presumes that since risk is proportional to dose, then dose can be used as a surrogate for risk. Additionally, since each dose is assumed to create some risk, those doses, and hence risks, are treated as additive. Therefore, with the absence of a threshold, risk can only increase with each dose, and this assumption applies to low as well as high doses. Importantly however, radiation protection practices (ICRP 1991) recognize the observation that different tissues respond differently to radiation, and, based only on the tissues actually exposed, individually contribute different fractions to the total risk of radiation. In practice, different tissue types are assigned tissue weighting factors (w T) that reflect their relative fractional contribution to the total cancer and non-cancer radiation risk. The w T for each tissue is held to be constant, independent of dose, since every tissue is assumed to obey a linear no threshold response. Another concept, also derived from observation and not the LNT hypothesis, is an assumed 2-fold reduction in the risk of a high dose/high dose rate exposure, if that exposure is received at low dose or low dose rate (ICRP 1991).
The dose limits established by various national bodies make no allowance for persons who may be at increased risk for genetic reasons, but simply assume that the limits provide adequate protection for all persons. This approach inherently assumes that the biological response to radiation of genetically cancer prone individuals is qualitatively similar to genetically normal individuals, and that a conservative approach to setting dose limits adequately compensates for any quantitative differences. However, since these assumptions have not been tested in humans, the concern arises that those risk predictions and hence dose limits may not be appropriate for genetically cancer prone individuals. If cancer prone humans were abnormally sensitive to low doses, such as those typical of public and most occupational exposures, then those persons would be at higher risk from such exposures.
Guidance for dealing with this concern in humans may be derived from the results of animal experiments. The TP53 gene functions in regulatory pathways for apoptosis, DNA repair and cell cycle delay, all processes considered to be critical for cellular responses to radiation damage, and hence for risk of both cancer and teratogenesis (Lee et al. 1994; Saintigny et al. 1999; Coates et al. 2003). Animals with TP53 defects are cancer prone (Harvey et al. 1993) but mice with constitutively activated TP53 are almost cancer free (Tyner et al. 2002). Importantly, the observations of increased cancer risk in mice with TP53 defects are paralleled in humans, where a single amino acid change in the p53 protein (proline for arginine at codon 72) has been shown to impair apoptosis (Dumont et al. 2003) and result in a 2.5 fold increase in cancer mortality (van Heemst et al. 2005). Interestingly, the increased cancer risk in humans with this mutation was accompanied by a 41% increase in survival in persons over the age of 85, a result again predicted by the previous similar mouse study (Tyner et al. 2002).
The present work reviews the radiation response of animals with defects in the TP53 gene, and compares those responses to the responses of animals with fully functioning TP53 genes. Two measures of risk are examined, carcinogenesis and teratogenesis.
DOSE ADDITIVITY, TP53 AND CARCINOGENESIS
Cancer is considered to be the most important risk associated with radiation exposure. If the LNT hypothesis is correct, sequential exposures to radiation should increase cancer risk for all types of exposures. Figure 1 shows a test of this concept in TP53 normal and heterozygous mice. Figure 1A shows that in TP53 normal mice exposed to two separate doses, 100 mGy at low dose rate, followed 24h later by exposure to 1 Gy, the latency for myeloid leukemia was increased as compared to a single 1 Gy exposure (Mitchel et al. 1999). While the frequency for myeloid leukemia was not different in the two cases, the additional 100 mGy exposure produced a protective effect, restoring about half of the life span otherwise lost due to the induction of this disease by the single 1 Gy exposure.

Cancer risk from sequential 60Co gamma radiation exposures. Panel A. The risk of myeloid leukemia in TP53 normal mice exposed to 1 Gy or to 100 mGy followed 24h later by 1 Gy. (Data from Mitchel et al. 1999). Open circles, Control mice without myeloid leukemia; Closed triangles, mice receiving 1 Gy that did not develop myeloid leukemia; Closed circles, mice exposed to 1 Gy that developed myeloid leukemia; Open squares, mice exposed to 100 mGy followed 24h later by 1 Gy that developed myeloid leukemia. Panel B. The risk of lymphoma in cancer prone TP53 heterozygous mice exposed to 4 Gy, or 10 or 100 mGy followed 24h later by 4 Gy. (Data from Mitchel et al. 2004). Open circles, mice not exposed to radiation: Closed circles, mice exposed to 4 Gy at high dose rate; Open squares, mice exposed to 10 mGy at low dose rate 24h prior to a 4 Gy exposure at high dose rate; Open triangles, mice exposed to 100 mGy at low dose rate 24h prior to exposure to 4 Gy at high dose rate.
Figure 1B shows the results of a similar experiment examining dose additivity in cancer prone TP53 heterozygous mice (Mitchel et al. 2004). Two separate exposures, consisting of a dose of 10 mGy at low dose rate followed 24h later by a dose of 4 Gy, increased lymphoma latency (without a change in frequency) in these cancer prone mice, compared to the single 4 Gy exposure alone. The additional 10 mGy exposure produced a protective effect that was qualitatively similar to that observed in the TP53 normal mice. However, in the TP53 heterozygous mice, an increased dose of 100 mGy prior to a 4 Gy dose did not produce a protective effect as was seen in the TP53 normal mice with these two exposures.
These results support one aspect of the risk assumptions used in radiation protection, that cancer proneness modifies only the quantitative and not the qualitative response to radiation. It indicates therefore, that the response of normal individuals, with suitable quantitative adjustment, might be used to predict radiation response and hence risk in cancer prone individuals. However, both results demonstrate failures in other assumptions and concepts of risk prediction. The concept of dose additivity, when at least one exposure is to a low dose at low dose rate, did not hold, and the concept that risk can only increase as dose increases was likewise not supported. A comparison of the two results however, shows the influence of TP53 on cancer risk. In both cases, a low dose at low dose rate was able to lower the risk associated with a high dose exposure but reduced TP53 function restricted the upper dose level at which radiation could provide this protection. Once past this upper dose threshold, increased dose could increase risk, as currently assumed.
DOSE ADDITIVITY, TP53 AND TERATOGENESIS
Exposure of a fetus to radiation during the period of organogenesis can result in malformations. In the murine fetus, a convenient measure of the severity of the risk of these malformations is tail length at birth, since the magnitude of the reduction in length reflects the extent of cell death from the radiation exposure. Figure 2 shows the results of a high dose and dose rate radiation exposure of pregnant mice during the period of tail formation (Mitchel et al. 2002). Female mice were bred to simultaneously contain fetuses that were TP53 normal, TP53 heterozygous and TP53 null. Figure 2A shows that the functional activity of TP53 had a large influence on the risk of radiation-induced tail malformation. Fetal mice with fully functional TP53 were at the highest risk of tail shortening. Reduced TP53 function in the heterozygous fetuses resulted in less risk, and the complete absence of TP53 function resulted in the least risk. Similarly, Wang et al. (1999) showed that susceptibility to radiation-induced apoptosis in the predigital regions and the resulting digital defects depended on Trp53 status, with TP53(+/+) mice the most sensitive, TP53(+/–) intermediate, and TP53(–/–) the most resistant. These results are consistent with the known importance of TP53 dependent apoptosis as a response to high doses and as a risk determinant for fetal malformations, but additionally show that a large portion of the risk can also be attributed to a TP53 independent process.

Radiation exposure and teratogenesis in fetal mice. (Data from Mitchel et al. 2002). Panel A. Tail shortening in fetal mice of varying TP53 function exposed to 4 Gy of gamma radiation delivered at high dose rate on gestational day 11. Panel B. Tail shortening in fetal mice of varying TP53 function exposed to 0.3 Gy 24h prior to a 4 Gy exposure of gamma radiation delivered at high dose rate on gestational day 11. Panel C. Tail shortening in fetal mice of varying TP53 function exposed to 0.3 Gy 24h prior to a 4 Gy exposure of gamma radiation delivered at high dose rate on gestational day 12. All Panels; Open symbols, control unirradiated fetal mice; Closed symbols, fetal mice exposed to 4 Gy; Shaded symbols, fetal mice exposed to 0.3 Gy 24h prior to 4 Gy exposure; Circles TP53 normal; Triangles, TP53 heterozygous; Squares, TP53 null.
At high doses and dose rates, the extent of the malformations in normal animals is known to be dose dependent, but exhibits an apparent threshold at about 30–50 cGy (Hossain et al. 1999). However, fetal irradiation with 2 Gy at 1.2 mGy/min was not teratogenic for Trp53(+/+) mice but was teratogenic for Trp53(–/–) mice (Kato et al. 2001). This indicates that the TP53 gene is indispensable for the threshold effect in the teratogenic risk of radiation at low doses or dose-rates.
These results demonstrate that the risk of fetal malformation as a result of a high dose exposure is highly dependent on TP53 function, and that in populations with varying TP53 function, dose is a poor surrogate for risk. In contrast to the concern of an increased risk of cancer related to cancer proneness, reduced TP53 function, and the associated cancer proneness, acts to reduce teratogenic risk at high dose rates but increase risk at low dose rates. This influence of TP53 could have interesting environmental implications. In an environment where pregnant wild mice may encounter a high dose rate exposure, the reduced teratogenic risk in fetal variants with reduced TP53 function would tend to increase the probability that such mice would be born with, for example, all feet and toes, increasing the probability that they would survive to successfully breed. Such a situation could put selective pressure for, rather than against, reduced TP53 function. Conversely, in a situation of low dose rate environmental exposure, the selective pressure would be reversed.
Aside from the varying risk of malformation associated with varying TP53 function, the LNT hypothesis would nonetheless predict that the inherent radiation risk associated with each level of TP53 function should increase proportionately with increasing dose. A test of this assumption of dose additivity for each TP53 state is shown in Figure 2B. Two separate doses, consisting of a dose of 0.3 Gy on gestational day 10 followed by a dose of 4.0 Gy on gestational day 11, produced effects that varied both qualitatively and quantitatively with TP53 function. Compared to the single 4.0 Gy exposure, the addition of another 0.3 Gy exposure did produce an increased risk of malformation in the TP53 null fetuses, as predicted by the LNT assumptions. In contrast however, the additional exposure reduced risk in the TP53 normal fetuses. A statistically significant, but much smaller protective effect was seen in the TP53 heterozygous fetuses. These results show that for fetal malformations, the concept of dose additivity, a fundamental assumption of risk prediction, depends entirely on the functional state of TP53, and only in the highly unusual case of a complete lack of TP53 function does the risk actually increase in accordance with the current dose additivity concept. The inappropriateness of the dose additivity concept and the importance of TP53 function in regulating teratogenic risk is further exemplified by an experiment where the same two exposures were each given one day later in gestation. Figure 2C shows those results. Since tail development was advanced by one day of gestation, the magnitude of the tail shortening by the single 4.0 Gy exposure was less than that seen when the exposure was given one day earlier (Figure 2A). However, the two-exposure protocol now produced different results from those seen when the same exposures were given one day earlier in gestation. Unlike the protective effects seen in TP53 normal and heterozygotes seen after the one day earlier exposures (Figure 2B), the additional dose had no effect in these more developed fetuses (Figure 2C). However, the additional dose now reduced the risk of malformation in the TP53 null fetuses, unlike the increased risk seen when the exposures were given earlier in gestation. These data emphasize that the concepts of dose additivity and increasing risk with increasing dose do not apply to fetal malformation risk, and that both the direction of the change and the final magnitude of the risk resulting from an additional dose is actually determined by a combination of gestational time and TP53 status.
COMBINED EFFECTS AND TP53
The TP53 gene controls biological processes that are important for the cellular response to a radiation exposure. These same processes can also be important for cellular responses to other stresses, and additionally, TP53 is known to regulate normal fetal developmental processes (Boreham et al. 2002a). As a consequence it could be expected that the net risk from other stresses combined with a radiation exposure may be modified by variations in TP53 gene function. Currently, radiation risk prediction does not include recognition of other risk-modifying factors, including genetic variations and non-radiation stresses.
One common stress in humans is fever, and heat is a known mild teratogen in both human (Graham et al. 1998; Martinez-Frias et al. 2001) and rodent fetuses (Germain et al. 1985; Boreham et al. 2002a) but its teratogenic effect is independent of TP53 status (Boreham et al. 2002a). Figure 3 shows the effect of the addition of a mild, short duration heat stress on the teratogenic risk of a high dose radiation exposure in murine fetuses with varying TP53 function (Boreham et al. 2002b). Exposure of the fetuses to radiation immediately after a heat stress on gestational day 11 amplified the teratogenic effect of radiation in fetuses lacking TP53 function, but had little or no influence in fetuses with full or partial TP53 function (Figure 3A). In contrast, delaying the radiation exposure by 24h to gestational day 12 resulted in a large increase in teratogenic risk for fetuses with either full or partial TP53 function, and a smaller increase in fetuses without TP53 function (Figure 3B).

Teratogenesis in fetal mice exposed to combined heat and radiation stress. (Data from Boreham et al. 2002b). Panel A. Tail shortening in fetal mice of varying TP53 function exposed to 4 Gy of gamma radiation delivered at high dose rate on gestational day 11, with or without a heat stress immediately prior to the radiation exposure. Panel B. Tail shortening in fetal mice of varying TP53 function exposed to 4 Gy of gamma radiation delivered at high dose rate on gestational day 12, with or without a prior heat stress on gestational day 11. Heat stress was at 40.5°C for 60 min. All Panels; Open symbols, control unirradiated fetal mice; Closed symbols, fetal mice exposed to 4 Gy; Shaded symbols, fetal mice exposed to heat stress at 40.5°C for 60 min either immediately or 24h prior to 4 Gy exposure; Circles TP53 normal; Triangles, TP53 heterozygous; Squares, TP53 null.
Heat stress can also modify the risk of cancer resulting from a high dose. In a result analogous to that shown in Figure 1A for a pre-exposure to a low dose of radiation in TP53 normal mice, exposure to a mild heat stress followed 24h later by exposure to 1 Gy increased the latency for myeloid leukemia as compared to the 1 Gy exposure alone. The magnitude of this heat-induced protective effect was also similar to the magnitude of the protection induced by the low dose radiation exposure. (Mitchel et al. 1999).
The current system of risk prediction using the LNT hypothesis and associated assumptions has no basis to allow for these combined effects with or without the influence of genetic variation.
SINGLE EXPOSURE, THRESHOLDS AND TISSUE WEIGHTING FACTORS
The data shown in Figures 1A and 1B supported the assumption that the radiation response of cancer prone individuals was qualitatively similar to that of normal individuals, even though the actual nature of the response to a low dose was opposite of that predicted by the LNT based dose additivity concept. TP53 heterozygous mice have a high spontaneous cancer frequency and cancers also appear much earlier than in TP53 normal mice (Harvey et al. 1993). They are therefore useful models to investigate risk prediction based on the LNT concept of a lack of a threshold for increased risk after a radiation exposure, and the associated concept of constancy of tissue weighting factors.
Figure 4 shows the effect of single low dose/low dose rate exposures on spontaneous cancer formation in the TP53 heterozygous mice (Mitchel et al. 2003). Figure 4A shows that a single low exposure had the same tumor latency increasing effect on spontaneous cancers as it did on radiation induced cancer in TP53 normal mice (Figure 1A) or TP53 heterozygous mice (Figure 1B). Also as before, the low dose did not alter tumor frequency. This observation again supports the current operational assumption that cancer proneness does not qualitatively alter radiation response. However, as was shown in Figure 1 for a low dose added to a high dose, a single low dose by itself (Figure 4A) did not increase risk, as predicted by the LNT hypothesis, and therefore risk was not proportional to dose. Since, in fact, risk decreased below the background risk in these cancer prone mice, Figure 4A shows that LNT hypothesis also fails at the level of the “no threshold” component for risk prediction. Clearly, a dose threshold exists for increased risk from a single radiation exposure. It is also interesting to note in Figure 4A that both 10 and 100 mGy single doses decreased spontaneous cancer risk in the TP53 heterozygous mice, indicating that the threshold for increased risk was above 100 mGy. This dose threshold for increased risk was higher than that seen for the same tumor type in the same animal genotype when the low dose was followed by a high dose (Figure 1B), indicating that for risk prediction in vivo, the threshold for increased risk depends not only on the initial dose but also on subsequent exposure levels. Since all these data support the concept that cancer proneness due to TP53 heterozygosity alters only the quantitative response to radiation, the data predict that a single exposure would be able to reduce spontaneous risk in TP53 normal mice at a dose higher than that in Trp53 heterozygous mice. That prediction remains to be tested.
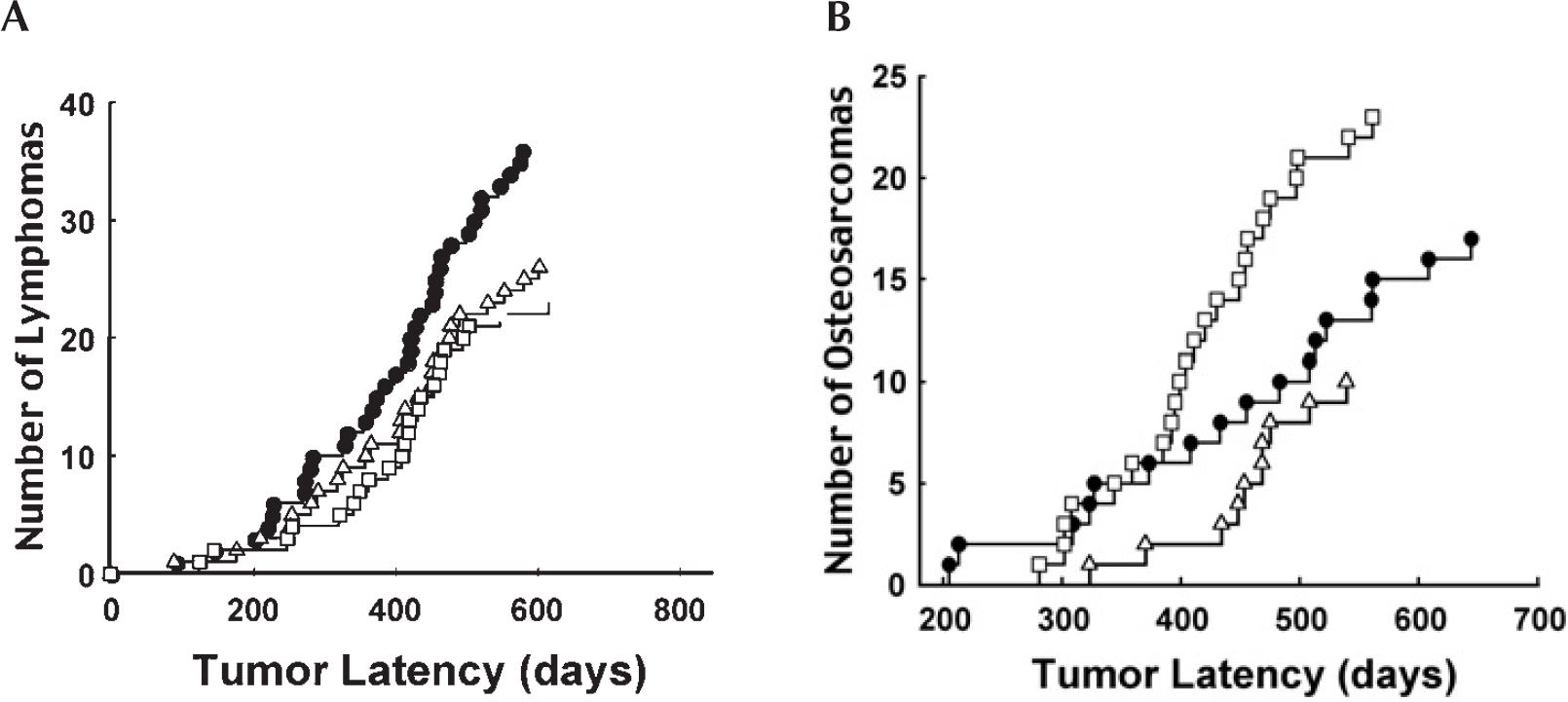
The effect of single low dose/low dose rate exposures on spontaneous cancer formation in TP53 heterozygous mice. (Data from Mitchel et al. 2003). Panel A. Spontaneous lymphoma formation in unexposed TP53 heterozygous mice (closed circles) and in mice with a single exposure to 10 (open triangles) or 100 mGy (open squares) of gamma radiation given at low dose rate to the mice at 8 weeks of age. Spontaneous lymphoma formation in unexposed TP53 normal mice is shown for comparison (open circles). Panel B. Spontaneous spinal osteosarcoma formation in unexposed TP53 heterozygous mice (closed circles) and in mice with a single exposure to 10 (open triangles) or 100 mGy (open squares) of gamma radiation given at low dose rate to the mice at 8 weeks of age.
In the TP53 heterozygous mice, a comparison can be made between the influence of a single radiation exposure on the spontaneous risk of lymphomas (Figure 4A) and on the spontaneous risk of osteosarcomas (Figure 4B). As for lymphomas, a single exposure to 10 mGy decreased risk by increasing tumor latency. However, unlike the observation for lymphomas in these animals, a dose of 100 mGy clearly exceeded the dose threshold at which risk increases. As for lymphomas, these data make the (as yet untested) prediction that this dose threshold for osteosarcomas will also be higher in TP53 normal animals.
The responses depicted in Figure 4 have implications for the LNT associated concept of using tissue weighting factors in risk prediction (ICRP 1990). Clearly, the data support the basic concept of tissue weighting factors, in that individual tissues differ in their quantitative response to radiation. However, equally clearly, the data do not support the concept of a constant w T value for any given tissue. At the dose point where risk neither increases nor decreases in a given tissue (for example, between 10 and 100 mGy for bone in the TP53 heterozygous mice), the w T for that tissue must equal zero; i.e. predict no contribution to the overall net risk in the animal. Above that threshold dose the w T would be positive (i.e. the tissue contributes positively to the net risk in the animal), as is the current practice. Below the threshold dose, the risk contribution from the specific tissue must be negative; i.e. there was a protective effect as the risk was below spontaneous risk. Correspondingly, the w T for that tissue should be negative, even though the overall net risk to the animal may be either positive or negative. These data driven considerations for tissue weighting factors represent a significant departure from the current approach for risk prediction at low doses.
An underlying assumption of the current system for risk prediction is the idea that biological effects can only occur in tissues that are actually exposed to radiation. However, it has been known for some time that radiation exposure of one site in the body can result in an antitumor effect for tumors at other sites, an effect termed the “abscopal effect.” This “action at a distance” has been shown to be dependent on functional TP53 (Camphausen et al. 2003), and is a direct indication that the effects of radiation are not limited to the exposed tissue. These observations suggest that the use of w T for risk prediction based on only the exposed tissue is incorrect.
DOSE RATE EFFECTS
An integral part of current radiation risk prediction is an assumed 2-fold reduction in the risk of an exposure received at low dose rate, compared to high dose rate. Such reductions in risk have been demonstrated many times after high dose exposures (ICRP 1991). The data shown in Figures 1,2,4, for both normal and reduced TP53 function indicate that low doses received at low dose rate reduce risk, but that the reduction takes the risk below the reference baseline. No matter what the positive risk of any high dose rate exposure might be, if the risk associated with a low dose rate exposure is protective (i.e. less than zero) then the dose rate reduction factor must be infinity rather than 2. The dose point at which the dose rate reduction factor becomes infinity must vary with both tissue type and TP53 status, in concert with the dose threshold (point of zero risk) under each situation.
RADIATION WEIGHTING FACTORS, SIEVERTS AND TP53
Ionizing radiation is broadly classified into high or low linear energy transfer (LET) radiation, based on energy deposition per unit track length. High LET radiation (for example, alpha radiation) is believed to have a greater risk per unit of absorbed physical dose (Gy) than low LET radiation (for example, gamma radiation). Radiation weighting factors (w R) are defined for high LET radiation to allow dose normalization, which in turn allows dose (i.e. risk) additivity and hence risk prediction on a unified basis. Physical doses (Gy) normalized by multiplication with w R are reported in sievert (Sv) units. Radiation weighting factors represent the ratio of the biological risk from high LET radiation exposure relative to the risk from low LET radiation exposure (where low LET radiation w R is defined as = 1). wR are assumed to be constant with dose and dose rate (ICRP 1991). However, the data shown in Figures 1,2,4 indicates that risk from a low LET radiation exposure varies as dose decreases, from increased risk, to no risk, to protection. This indicates that w R for low LET radiation is not constant with dose and cannot be assigned a w R=1 for all doses. At the low LET dose threshold for increased risk, w R must be zero and below that threshold must be a negative number. Consequently w R values for high LET radiation have no meaning when referenced to a low LET radiation dose where there is no, or negative, risk. Since animals with either normal or reduced TP53 function respond qualitatively in the same way, the use of w R at low doses at or below the “no risk” threshold in either situation would not be meaningful. Because, however, the threshold dose points are quantitatively different in the two cases of differing TP53 function, the low dose point at which the use of w R becomes meaningless will differ in those two situations. As a consequence, the data indicate that risk prediction based on dose normalization to sievert units is also meaningless below these dose points.
CORRELATION OF IN VIVO WITH CELLULAR DATA
The above observations of the influence of TP53 on the effects of low dose exposure in vivo are consistent with observations of low dose responses in cells. Low doses of low-LET radiation induce protective effects and those induced responses have been tightly conserved throughout evolution, suggesting that they are basic responses critical to life (Mitchel, 2005). Induction of DNA repair, suppression of apoptosis and protection against radiation and other DNA damaging agents are central features of this induced response. These responses have been shown to impact on cancer risk, even as measured in vitro, since low doses of low LET radiation have been shown to reduce the risk of spontaneous neoplastic transformation in both rodent (Azzam et al.1996) and human cells (Redpath and Antonionio, 1998) in culture. In G0 human fibroblasts, high dose rate exposure induced TP53 phosphorylation and H2AX foci in a dose dependent manner, but low dose rate exposure resulted in virtually no foci or TP53 phosphorylation, suggesting that the efficient repair of DNA double strand breaks induced by low dose rate exposure is linked to TP53 response (Ishizaki et al. 2004). The anti-apoptotic adaptive responses seen in vivo have been shown to be related to regulation of TP53 in the spleens of exposed mice. High dose and dose rate exposure stimulated TP53 accumulation and apoptosis, but TP53 accumulation was suppressed, along with apoptosis, after a pre-irradiation at low dose rate (Takahashi et al. 2001). While TP53 has been shown to be inducible by very low doses in mice, with increasing response with increasing dose in both radiation-resistant and radiation-sensitive tissues (MacCallum et al. 2001), the response was heterogeneous between cells, suggesting a cellular basis for the differential tissue response and tissue specific thresholds noted here.
CONCLUSIONS
The currently accepted set of assumptions used for the prediction of risk from a radiation exposure makes no allowance for genetic differences between individuals or for radiation combined with other stresses. One particular gene, TP53, has been shown to be important in the biological processes that control radiation responses for cancer formation and birth defects. Risk estimate assumptions concerning a linear response with dose, dose additivity, lack of thresholds and dose rate reduction factors all appear incorrect at low doses. TP53 functional variations can further modify radiation risk from either high or low doses, or risk from radiation exposures combined with other stresses, and those modifications can result in both quantitative and qualitative changes in risk, an outcome not accommodated in the current system of radiation risk prediction.
Footnotes
ACKNOWLEDGMENTS
This work was supported by Atomic Energy of Canada Limited.