
Abstract
A stochastic two-stage cancer model with clonal expansion was used to investigate the potential impact on human lung cancer incidence of some aspects of the hormesis mechanisms suggested by Feinendegen (Health Phys. 52 663–669, 1987). The model was applied to low doses of low-LET radiation delivered at low dose rates. Non-linear responses arise in the model because radiologically induced adaptations in radical scavenging and DNA repair may reduce the biological consequences of DNA damage formed by endogenous processes and ionizing radiation. Sensitivity studies were conducted to identify critical model inputs and to help define the changes in cellular defense mechanisms necessary to produce a lifetime probability for lung cancer that deviates from a linear no-threshold (LNT) type of response. Our studies suggest that lung cancer risk predictions may be very sensitive to the induction of DNA damage by endogenous processes. For doses comparable to background radiation levels, endogenous DNA damage may account for as much as 50 to 80% of the predicted lung cancers. For an additional lifetime dose of 1 Gy from low-LET radiation, endogenous processes may still account for as much as 20% of the predicted cancers (Fig. 2). When both repair and scavengers are considered as inducible, radiation must enhance DNA repair and radical scavenging in excess of 30 to 40% of the baseline values to produce lifetime probabilities for lung cancer outside the range expected for endogenous processes and background radiation.
INTRODUCTION
There are biological responses to a variety of chemical and radiological agents that may be U-shaped rather than LNT (Calabrese and Baldwin 2003). Azzam et al. (1996) demonstrated that low doses (1–100 mGy) of γ-rays, when delivered at 2.4 mGy min−1, reduced the neoplastic transformation frequency in C3H 10T1/2 cells to a rate three- to four-fold below the spontaneous transformation frequency. This has been confirmed in human-hybrid cell systems (Redpath and Antoniono 1998, Redpath et al. 2001, 2003a, 2003b). In animals in vivo, low doses have been shown to reduce tumor frequency (Ishii et al. 1996) and increased both radiation-induced and spontaneous tumor latency (Mitchel et al. 1999, 2003). These results demonstrate that low-dose exposures may induce processes, such as adaptations in DNA repair processes (Le et al. 1998, Ye et al. 1999), radical scavenging (Yamaoka et al. 1991, Yukawa et al. 1999) or apoptosis (Bauer 1995, 1996, 2000) that reduce the overall rate of cell transformation. In addition to the in vitro and animal in vivo studies, a review of human epidemiological studies suggests that protracted exposure to low doses of low-LET radiation does not appear to cause lung cancer and a reduction of the natural cancer incidence level may even be evident (Rossi and Zaider 1997).
In an earlier study (Schöllnberger et al. 2004), the concept of radiologically induced adaptations that also prevent and repair endogenous (oxidative) DNA damage was developed using a deterministic 3-stage lung cancer model. In this work, we develop methods of incorporating these mechanisms into a stochastic two-stage cancer model. In a deterministic model the hazard rate increases indefinitely while in a stochastic cancer model it levels off at advanced ages (Heidenreich and Hoogenveen 2001, Chen 1993). The trends predicted by stochastic models are consistent with observations suggesting that lung cancer incidence and mortality rates increase with age, but start to level off at advanced age and (for many cancers) decline at very high ages (see e.g., Benson 1996, Herrero-Jimenez et al. 2000).
The aim of the current study is to define the magnitude of the changes in DNA damage repair and enzymatic radical scavengers that would be required to produce a lifetime probability for lung cancer that deviates from a linear no-threshold (LNT) type of response. Studies are presented for hypothetical exposure scenarios in which individuals are exposed to low doses rates of low-LET radiation in addition to natural background radiation. The reported studies are most appropriate when high-LET radiations, such as α particles from radon progeny, are a minor component of the natural background radiation. Although continuous exposure to low dose rates of low-LET radiation are not typical of many environmental exposure settings, this type of exposure setting is a reasonable facsimile of the types of exposures that may be encountered by workers in nuclear power plants or other radiation facilities. The reported studies are the first to consider the effects that radiation-induced adaptations in cellular defense mechanisms have on predictions of lung cancer incidence made with a stochastic multistage model. The approach developed in this work is an important first step in developing new models and methods to describe dose-response relations that deviate from the LNT response models that are widely used for radiation protection purposes.
MATERIALS AND METHODS
Key aspects and the mathematical properties of stochastic two-stage cancer models have been extensively discussed in the literature (Moolgavkar 1979, Moolgavkar and Venzon 1979, Moolgavkar and Knudson 1981, Moolgavkar and Luebeck 1990, Tan 1991, Little 1995, Heidenreich 1996, Heidenreich et al. 1997a, 1997b). Figure 1 shows an idealized schematic of the two-stage model. In this model, normal cells (N-cells) are converted to initiated cells (I-cells) at the stochastic rate μ1. Initiated cells complete mitosis and produce two progeny cells at stochastic rate α and die or differentiate at stochastic rate β. In addition, initiated cells can also divide into one initiated and one malignant cell (M-cell) at stochastic rate μ2. Malignant cells develop into a detectable tumor mass after a non-stochastic lag time, t lag , for which a value of 5 years is used (Leenhouts 1999). The net clonal expansion rate of initiated cells, α-β, is assumed to be independent of dose and dose-rate. In the model, radiation and endogenous processes can damage the DNA of target cells in the lung. Some fraction of the misrepaired or unrepaired DNA damage induces genomic instability and leads to the accumulation of initiated and malignant cells.

Conceptual overview of the two-stage cancer model with clonal expansion. See the text for explanation of the symbols.
To examine the potential significance of radioprotective mechanisms, dose and dose rate dependent DNA repair and radical scavenging phenomena are incorporated into the model. Changes in DNA repair and radical scavenging with dose rate (and hence dose) are modeled using dimensionless scaling-functions, denoted G and F, respectively. For values of G and F greater than one, radiation is less effective at inducing genomic instability and cell transformation (i.e., reduces the rate μ1). Rate μ1 is parameterized as follows (Schöllnberger et al. 2004)

Here, Ω is the probability that a mutation formed at a random location in the DNA induces genomic instability by modifying the expression or function of a critical gene. φ i is the probability the ith type (simple or complex) of lesion is misrepaired. Σ i endo is the expected number of ith type lesion created by endogenous processes (cell−1 year−1), and Σ i rad is the expected number of ith type lesion created by radiation (mGy−1 cell−1), the dose rate, Ḋ, is the total delivered dose divided by a lifespan of 75 years. Both, G(Ḋ) and F(Ḋ), have the following form

A, B and C are adjusted so that G and F take on modified Gaussian forms. For Ḋ m two different values were used: 2.67 and 4 mGy yr−1. This results in maxima of G(Ḋ) and F(Ḋ) at these indicated dose-rates, respectively at the associated lifetime doses of 200 and 300 mGy (Schöllnberger et al. 2004). This means that the maximum adaptive protection due to changes in the misrepair probability, φ i , and the radical scavenging capacity of a cell will occur at dose rates of 2.67 and 4 mGy yr−1. These values were chosen to reflect different hypothetical exposure scenarios where doses of 200 or 300 mGy of low-LET radiation are delivered at low dose rates in addition to background radiation. The 200 to 300 mGy delivered in these exposure scenarios correspond to the extra dose of radiation a worker might receive while working in, for example, a nuclear power plant.
The exact tumor incidence formula in closed form developed for the stochastic two-stage model by Kopp-Schneider et al. (1994) has been applied. A lag-time was introduced into their formula that was then used to calculate the lifetime probability for lung cancer. In another study (Schöllnberger et al. 2004), a deterministic 3-stage cancer model was used to identify model parameters consistent with the ICRP-derived risk coefficient (0.85 × 10−2 Sv−1) for fatal lung cancer (ICRP 1991). A similar methodology was used to identify parameters for the stochastic cancer model. All parameter values were taken from Table 1 in Schöllnberger et al. (2004) except for the following: φ cl = 0.4937, Σ cl endo = 0 cell−1 yr−1, μ2 = 10−9 yr−1, α = 0.05 yr−1, β = 0 yr−1. The first three values were found by fitting the model solution to the ICRP-derived values for the lifetime probability for lung cancer at lifetime doses of 75, 225, and 1000 mGy. The value for α reflects the difference of earlier applied values for the mitotic rate and the cell death and differentiation rate for initiated cells (Schöllnberger et al. 2004). With β = 0, α is the net clonal expansion rate of initiated cells.
RESULTS
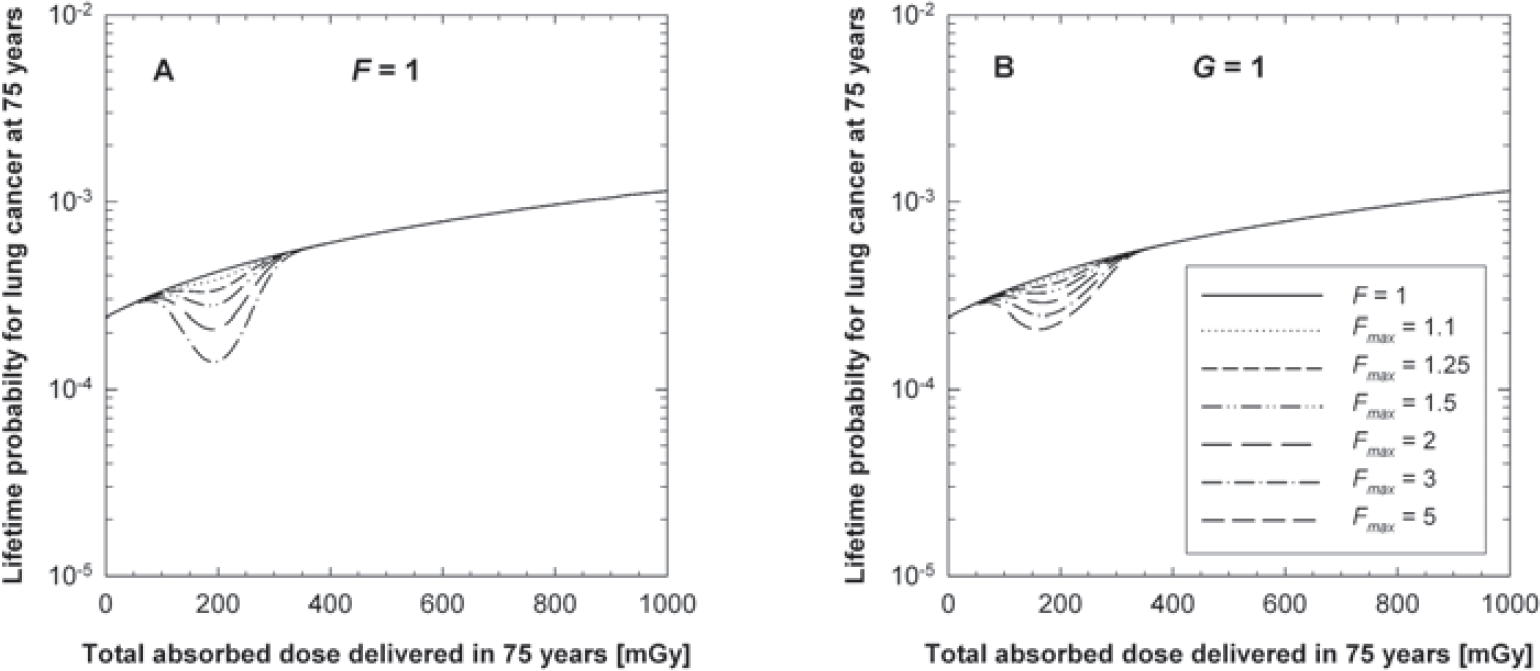
Fig. 2 shows the lifetime probability for lung cancer at an age of 75 years versus the total absorbed dose delivered in the same time span. The corresponding dose-rates range from 0 to 13.33 mGy/yr. The solid and dotted curves show the predicted lifetime probability of lung cancer obtained with the stochastic and deterministic 3-stage cancer model, respectively. The dashed curve shows the predicted lifetime probability for radiation alone (i.e., Σ endo complex = Σ endo single = 0). Fig. 3 illustrates the effects that radiation-induced adaptations in DNA damage repair (panel A) and radical scavenging (panel B) may have on the lifetime probability for lung cancer at an age of 75 years. The x-axis in Fig. 3 gives the lifetime dose from low-LET radiation. The results in Fig. 3 refer to Ḋ m = 2.67 mGy yr−1, i.e., we assumed that maximum gene induction occurs at 2.67 mGy yr−1 from low-LET radiation which is delivered in addition to a very low lifetime dose of background radiation. Fig. 4 shows the combined effects for two values of Ḋ m (2.67 and 4 mGy yr−1) and with various maxima in G and F.
DISCUSSION
For doses comparable to background radiation levels, the results shown in Figure 2 suggest that endogenous DNA damage accounts for as much as 54 to 78% of the predicted lung cancers. For a lifetime dose of 1 Gy, endogenous processes may still account for as much as 21% of the predicted cancers (Fig. 2). The shape of the lifetime probability of lung cancer is very similar for both the stochastic and deterministic cancer models (Fig. 2). Sensitivity studies highlight the importance of including endogenously formed DNA damage in estimates of low-dose cancer incidence levels (Fig. 3). The results in Fig. 3, panel A, suggest that radiation must induce changes in DNA repair of at least 50% (G > 1.5) of the baseline value in order to produce cumulative probability levels for lung cancer outside the range expected for endogenous processes and background radiation. For scavengers (panel B) the changes must be at least 200% (F > 2) to lead to significant deviations in the dose-response curves for lifetime probability for lung cancer.

Contribution to the lifetime probability for lung cancer of DNA damage formed by endogenous processes and ionizing radiation. Protective effects are not included in this set of calculations (i.e., F = G = const. = 1). Solid curve: stochastic cancer model with DNA damage formed by ionizing radiation and endogenous processes. Dotted curve: deterministic 3-stage cancer model with DNA damage formed by ionizing radiation and endogenous processes (Schöllnberger et al. 2004). Dashed curve: stochastic model without any endogenous DNA damage (Σ endo complex = Σ endo single = 0). The vertical dotted lines indicate the typical dose range expected from naturally occurring radiation sources, i.e., background radiation. The lower dose bound is set at 75 mGy (1 mGy yr−1), and the upper dose bound is set at 225 mGy (3 mGy yr−1).
Effects of cellular adaptations in DNA repair (panel A) and enzymatic radical scavenging (panel B) on the lifetime probability of lung cancer at an age of 75 years for various doses of low-LET radiation which is delivered in addition to a very low lifetime dose of background radiation. Ḋ m = 2.67 mGy yr−1, i.e., it is assumed that maximum gene induction occurs at 2.67 mGy yr−1 from low-LET radiation which corresponds to a lifetime dose of 200 mGy.
Depending on the chosen values used for Ḋ m , the model predicts effective cancer incidence thresholds from 300 mGy (4 mGy yr−1 for 75 years) up to approximately 375 mGy (5 mGy yr−1 for 75 years) (Fig. 4). The results in Fig. 4 suggest that radiation must induce changes in radical scavenging and DNA repair greater than about 30 or 40% (F and G > 1.3 to 1.4) of the baseline values in order to produce cumulative probability levels for lung cancer outside the range expected for endogenous processes and background radiation. Some experiments suggest that, in non-dividing cells, DNA repair remains at some baseline level until a threshold dose is exceeded (Marples et al. 2002, Oudalova et al. 2002, Rothkamm and Löbrich 2003, Zaichkina et al. 2003). Fig. 4, panel B, shows the response expected if DNA repair and radical scavenging remain at a baseline level until a threshold dose on the order of 150 mGy is exceeded. Others, however, found that doses as low as 1 mGy induce the full adaptive response (Broome et al. 2002). The 150 mGy threshold for the induction of adaptive protection is a model prediction for a Ḋ m -value of 4 mGy yr−1. Model predictions such as those shown in Fig. 4 appear counter to some available epidemiological data for solid cancers (Pierce and Preston 2000, Brenner et al. 2003) but find support by others (Rossi and Zaider 1997). Our predictions are also consistent with the findings of Azzam et al. (1996), Redpath and Antoniono 1998, Redpath et al. 2001, 2003a, 2003b) for neoplastic transformation in vitro. In vitro assays of neoplastic transformation have been shown to have relevance to radiation carcinogenesis in vivo (Little 1989). In addition, Redpath et al. (2001) found a remarkable similarity between the relative risks calculated from their in vitro studies to those from various epidemiological studies (Thompson et al. 1994), particularly for breast cancer (Miller et al. 1989) and leukemia (Preston et al. 1994, Little et al. 2000).

Predicted shapes of the lifetime lung cancer probability curves when both cellular defense mechanisms (inducible DNA repair and radical scavenging) are included in the model. Panel A: Ḋ m = 2.67 mGy yr−1; panel B: Ḋ m = 4 mGy yr−1.
The 30 to 40% change in DNA repair and scavenging used in the modeling studies are consistent with the results of several experiments. Le et al. (1998) report an app. 2-fold enhanced repair response for radiation-induced DNA base damage. The increased rate of lesion removal was caused by a 0.25 Gy dose of γ-radiation. Azzam et al. (1994) showed that a dose of 0.5 Gy of γ-radiation (0.56 mGy/min) enhances the rate of DNA repair in human fibroblasts by app. 30%. The same group also showed that even 1 or 10 mGy doses (2.4 mGy/min) lead to the same significant reduction of the neoplastic transformation frequency in C3H 10T1/2 cells below the spontaneous transformation frequency than a 100 mGy dose (Azzam et al. 1996). This indicates that a low dose of 1 mGy can also cause adaptive protections to a very similar degree as the one reported for 0.5 Gy. Yukawa et al. (1999) demonstrated a transient increase in liver cytosolic radical scavenging ability after an in vivo low dose irradiation of rats. The maximum level of induction (app. 20%) was found around 5 to 10 cGy (35 cGy/min) of X-rays (Yukawa et al. 1999). It should be emphasized that the dose rates applied in these experiments are considerably higher than those considered in the simulation studies reported in this work. Additional experimental work for lower dose rates is desirable.
To better model the types of radiation exposures encountered in the workplace, the proposed model needs to be extended so that time-dependent changes in DNA repair and radical scavenging caused by a specified dose of low-LET radiation delivered in a few minutes or hours can be simulated. The current approach is limited to low doses of low-LET radiation delivered at very low dose rates (i.e., dose rates comparable to background radiation levels). Methods to incorporate cellular defense mechanisms into biologically based neoplastic transformation models (Scott et al. 2003, 2004) and cancer models (Bogen 2001) are important to future developments in radiation protection. Experiments by Bauer (1995, 1996, 2000) and Belyakov et al. (2001, 2002, 2003) suggest that apoptosis and cell differentiation may be important radioprotective mechanisms and additional work is needed to integrate these and other phenomena into multi-stage cancer models.
CONCLUSIONS
Endogenous DNA damage should be considered in estimation of the risks from low doses of ionizing radiation. Our studies suggest endogenous damage may contribute significantly to the induction of lung cancer even for lifetime doses as high as 1 Gy.
Based on a stochastic two-stage lung cancer model that considers inducible DNA repair and radical scavenging together, radiation must induce changes in repair and scavenging greater than 30 or 40% of baseline values in order to produce significant deviations in the dose-response curves for the lifetime probability for lung cancer.
If only radical scavenging is considered, radiation must induce changes of at least 200% of the baseline scavenger activity in order to produce cumulative probability levels for lung cancer outside the range expected for endogenous processes and background radiation.
If only DNA repair is considered, radiation must induce changes of at least 50% of the baseline repair value in order to produce cumulative probability levels for lung cancer outside the range expected for endogenous processes and background radiation. DNA repair effects are predicted to have a larger impact on lung cancer incidence than radical scavenging.
The model neglects many other biological processes that are potentially important in low dose radiobiology, and additional work is needed. The proposed model also needs to be extended to other types of exposure conditions, such as low doses of low-LET radiation delivered at high dose rates.
Footnotes
ACKNOWLEDGEMENTS
The authors thank Dr. Marco J. Brugmans, RIVM, and Dr. Annette Kopp-Schneider, German Cancer Research Center, for fruitful discussions. We also wish to thank the journal referees for their valuable criticism. This work was supported by a Marie Curie Individual Fellowship (EC Contract No. FIGH-CT-2002-50513), by a Marie Curie European Reintegration Grant within the 6th European Community Framework Program (EC Contract No. MERG-CT-2004-006610) and in part by the ‘RISC-RAD’ project EC Contract No. FI6R-CT-2003-508842 and by the Austrian Science Foundation FWF (project P18055-N02). We also acknowledge support by the Low Dose Radiation Research Program, Biological and Environmental Research (BER), U.S. Department of Energy, Grant Nos. DE-FG02-03ER63541 and DE-FG02-03ER63665. We also acknowledge support from Atomic Energy of Canada Limited.