
Abstract
Mammalian cell-free systems are very useful for the biochemical and structural study of nuclear disassembly and assembly. Through experimental manipulations, the role of specific proteins in these processes can be studied. Recently, we intended to examine the involvement of integral and peripheral inner nuclear membrane proteins in nuclear disassembly and assembly. However, we could not achieve proper disassembly when isolated interphase HeLa nuclei were exposed to mitotic soluble extracts obtained from the same cell line and containing cyclin B1. Homogenates of synchronized mitotic HeLa cells left to reassemble their nuclei generated incomplete nuclear envelopes on chromatin masses. Digitonin-permeabilized mitotic cells also assembled incomplete nuclei, generating a lot of cytoplasmic inclusions of inner nuclear membrane proteins as an intermediate. These results were therefore used as a basis for a critical evaluation of mammalian cell-free systems. We present here evidence that cell synchronization itself can interfere with the progress of nuclear assembly, possibly by causing aberrant nuclear disassembly and/or by inducing the formation of an abnormal number of mitotic spindles.
Keywords
T
Nuclear lamins are type V intermediate filament proteins (Aebi et al. 1986). They form the nuclear lamina, an insoluble protein meshwork situated between the inner nuclear membrane and the chromatin, but are also present throughout the nucleus (Goldman et al. 1992; Bridger et al. 1993). The nuclear lamina provides support and strength to the NE (reviewed by Hutchison et al. 2001) and acts as a framework for attachment and organization of chromatin. Lamins have been subdivided into types A and B based on their molecular sequences (reviewed by Nigg 1992). The expression of lamin types varies according to cell lines. For example, B-type lamins seem to be expressed in all cell lines, whereas A-type lamins are absent from early developing embryos and undifferentiated or rapidly proliferating cells (Guilly et al. 1987, 1990; Paulin-Levasseur et al. 1996; Lin and Worman 1997; Worman and Courvalin 2000). Other proteins may be considered as non-lamin components of the lamina, such as otefin (Harel et al. 1989; Padan et al. 1990), young arrest (YA) (Lin and Wolfner 1991), circumferin (Chaly et al. 1984), and statin (Wang 1985).
The number of known proteins specifically associated with the INM is growing, especially because of the interest in such proteins as causes of human genetic diseases. They all possess at least one transmembrane domain and are associated with lamins and/or DNA. Because of these associations, they are believed to be crucial in chromatin organization and in the processes of nuclear disassembly and assembly. Integral INM proteins include lamin B receptor (LBR) (Worman et al. 1988, 1990), lamina-associated polypeptides (LAPs)1 (Senior and Gerace 1988), LAP2 (Foisner and Gerace 1993), emerin (Bione et al. 1994), MAN1 (Paulin-Levasseur et al. 1996; Lin et al. 2000), nurim (Rolls et al. 1999), SUN proteins (Dreger et al. 2001; Hodzic et al. 2004), and some isoforms of nesprin (Zhang et al. 2001). MAN1, LAP2, and emerin share a region of ∼40 residues called the LEM domain. The only known integral membrane proteins of pore domains are gp210 (Gerace et al. 1982; Wozniak et al. 1989) and POM121 (Hallberg et al. 1993). These proteins link the NPCs to the NE, which actively control the passage of macromolecules to and from the nucleus (reviewed in Vlcek et al. 2001).
Mitosis in higher eukaryotes involves complete disassembly and reassembly of the nucleus, referred to as “open mitosis.” Disassembly would occur in three main independent steps (Newport and Spann 1987): chromosome condensation, nuclear lamina depolymerization, and NE breakdown (NEBD). The last step would define the end of prophase and the beginning of prometaphase. Disassembly of the nuclear lamina correlates with its hyperphosphorylation, elicited by the cyclin B1-CDK1, previously known as the M-phase promoting factor (MPF), and by protein kinase C (PKC) (Peter et al. 1990; reviewed in Buendia et al. 2001). A number of integral and peripheral NE proteins, including LBR, LAP2β, LAP2α (Dechat et al. 1998), and emerin, are also believed to be phosphorylated at prophase by kinase cyclin B1-CDK1. Phosphorylation is thought to abolish the structural protein–protein integrity of the NE, promoting membrane release from chromatin. The release of NPCs also seems to be an important step in nuclear disassembly, leading to a fenestration of the NE before NEBD (Collas 1998; Terasaki et al. 2001).
At the onset of prophase, microtubules cause two symmetrical indentations on antidiametric sites of the NE where the centrosomes are located (Pawaletz and Lang 1988). Microtubule bundles are also found deep within NE invaginations (Georgatos et al. 1997). They have been shown to facilitate NEBD by literally tearing the NE open (reviewed by Burke and Ellenberg 2002). Amazingly, NEBD and fragmentation of the nuclear lamina can still occur in the absence of microtubules, as in nocodazole-treated cells or cell-free systems. However, the mode of nuclear disassembly of nocodazole-treated cells differs significantly from controls. The fragmentation pattern of lamin B is more rapid, and no nuclear indentations are present (Georgatos et al. 1997).
As mitosis progresses, reassembly of the nucleus is characterized by dephosphorylation through the activity of phosphatases (reviewed by Earnshaw and Pluta 1994). Assembly of the NE is also a stepwise process that starts in anaphase and is completed in early G1. Proteins start being targeted to chromosomes in anaphase A and B. As nuclear envelope–derived vesicles attach to chromosomes, they must fuse to enclose them. NPCs also resume their location sequentially, and the nuclear lamina continues to assemble, resulting in a functional nucleus. After the nucleus is enclosed, it enlarges as the NE and lamina grow to let the chromatin decondense and permit the formation of intranuclear structures, such as the nucleolus (Gant and Wilson 1997).
Many nuclear proteins have been shown to be essential in nuclear assembly. Experiments in Xenopus cell-free systems with truncated LAP2β proteins indicated that lamina assembly and membrane–chromatin attachment may be mediated by these proteins (Gant et al. 1999). Furthermore, it has been reported that LAP2α fragments can block assembly of nuclear membranes and lamins A/C in vitro (Vlcek et al. 2002).
In vitro systems of nuclear disassembly and assembly are especially attractive for structural and biochemical studies of mitosis. The most common cell-free system takes advantage of cellular mitotic extracts of Xenopus laevis (Lohka and Masui 1983; Newport and Spann 1987). However, because this system is amphibian and embryonic, there could be major differences from what occurs in somatic mammalian cells. Mammalian somatic cells have limited NE precursors, and some proteins involved in nuclear disassembly and assembly have isoforms that are differentially expressed during development.
A limited number of studies have been carried out on mammalian cell-free systems. Suprynowicz and Gerace (1986) disassembled nuclei isolated from Chinese hamster ovary (CHO) cells using a postribosomal supernatant from the same cells in metaphase. They observed prophase/prometaphase-like changes in the isolated nuclei, including chromosome condensation, NEBD, and lamina disassembly. More recently, cell-free nuclear disassembly experiments have been performed on human cells (Collas et al. 1999; Martins et al. 2000). Using a HeLa cell-free system, the function of some nuclear proteins in disassembly were blocked by introducing antibodies into lysolecithin-permeabilized isolated nuclei (Collas et al. 1999). Cell-free assembly systems were also developed, exploiting homogenates of metaphase CHO cells maintained in an appropriate physiological buffer (Burke and Gerace 1986; Burke 1998). As time elapses during these assembly assays, MPF is inactivated, and nuclear envelopes reform around chromosome clusters. The function of NE proteins in assembly can be blocked by adding specific antibodies to the homogenate (Burke and Gerace 1986). A novel in vitro assembly assay was also developed by Kourmouli et al. (2001), where nocodazole-synchronized HeLa cells are preincubated in low concentrations of digitonin to permeabilize the plasma membrane, allowing the addition of exogenous elements to the system.
These cell-free systems seemed quite promising, but after multiple attempts without and with modifications, they proved to be unreliable. Many difficulties were encountered in working out efficient assays. For disassembly, we were unable to obtain a functioning system using published protocols. Reactions were repeated many times and modified in many ways, but to no avail. As for assembly, mitotic cell homogenates and digitonin-permeabilized mitotic cells turned out to give questionable results and generally incomplete nuclear envelope reformation. Hence, this report essentially conveys a critical reassessment of mammalian cell-free systems of nuclear disassembly and assembly based on experimental evidence.
Materials and Methods
Materials
General biochemical and chemical supplies were purchased from BDH (Ville St-Laurent, Quebec, Canada) and Sigma (St Louis, MO).
Cell Culture
HeLa cells (CCL 2) were obtained from the American Type Culture Collection (ATCC; Manassas, VA), whereas CHO cells were kindly provided by Dr. Marc Ekker (Department of Biology, University of Ottawa, Ottawa, Ontario, Canada). HeLa cells were cultured in Eagle's MEM (Gibco BRL; Burlington, Ontario, Canada) supplemented with 10% FBS (Gibco BRL) and antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin; Gibco BRL), whereas CHO cells were grown in DMEM (Gibco BRL) supplemented with FBS and antibiotics. Cells were maintained at 37C in a humidified atmosphere with 5% CO2.
Cell Synchronization and Harvesting
Cells were grown up to 90% confluence (plated at a density of 1.5 × 104 cells/cm2 and grown for 48 hr or at 7.5 × 103 cells/cm2 and grown for 72 h) and either left untreated and asynchronous or treated for 18 hr with nocodazole (1 μM from a 3.3 mM stock in DMSO; Sigma) to arrest cells in mitosis. Untreated cells were washed in cell culture PBS (cPBS; 137 mM NaCl, pH 7.0, 8 mM Na2HPO4, 1.5 mM KH2PO4, 2.7 mM KCl) and detached in trypsin/EDTA (0.025% trypsin, pH 7.4, 1 mM EDTA, in cPBS), whereas cells synchronized with nocodazole were collected by the shake-off technique (Tobey et al. 1967). Subsequently, cells were recovered by centrifugation at 500 × g for 6 min.
Disassembly Systems
Mitotic Cell Extracts for Disassembly Assays. Cell extracts were obtained as previously described (Collas et al. 1999). Mitotic HeLa cells were washed in ice-cold PBS (130 mM NaCl, pH 7.0, 5 mM Na2HPO4, 1.5 mM KH2PO4) and centrifuged at 500 × g. The pellet was washed in 20 volumes of ice-cold lysis buffer [20 mM HEPES, pH 8.2, 5 mM MgCl2, 10 mM EDTA, 1 mM dithiothreitol (DTT), 20 μg/ml cytochalasin B, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin A] and centrifuged at 800 × g for 10 min. In some preparations, EDTA was replaced with EGTA. The pellet was resuspended in 1 volume of lysis buffer and incubated for 30 min on ice. Homogenization was achieved by sonicating (Braun-Sonic 2000; B. Braun Instruments, Allentown, PA) twice for 2 min on ice. The homogenate was centrifuged at 10,000 × g for 15 min at 4C, and the supernatant was centrifuged at 200,000 × g (45,000 rpm) for 3 hr at 4C in a Beckman SW55 Ti rotor (Beckman Coulter; Fullerton, CA) with adaptors for 0.8-ml Ultra-Clear tubes. Clear supernatant, later referred to as the soluble mitotic extract, was separated into aliquots and snapfrozen in liquid nitrogen to be stored at −80C. Both low-speed and high-speed pellets were kept in sample buffer (2% SDS, 10% glycerol, 10 mM Tris-base, pH 6.8, 25 mM mercapto-ethanol, 0.005% bromophenol blue) for analysis.
An alternative method was devised from a protocol by Burke (1998). After incubation in lysis buffer, cells were homogenized on ice by 25 strokes of a tight-fitting pestle in a dounce homogenizer. The resulting homogenate was centrifuged at 100,000 × g (32,500 rpm) in a SW55 Ti Beckman rotor with 0.8-ml tube adaptors at 4C for 30 min. The supernatant, which is the soluble mitotic extract, was frozen as in the previous method, and the pellet was kept in sample buffer for analysis.
Preparation of Nuclei for Disassembly Assays. Interphase nuclei were isolated as described by Collas et al. (1999). Un-synchronized confluent HeLa cells were harvested, washed in PBS, and centrifuged at 400 × g. The pellet was resuspended in 20 volumes of ice-cold nuclear isolation buffer (buffer N; 10 mM HEPES, pH 7.5, 2 mM MgCl2, 250 mM sucrose, 25 mM KCl, 1 mM DTT, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin A) containing 10 μg/ml cytochalasin B and incubated for 30 min on ice. Cells were homogenized on ice with 150 strokes of a tight-fitting glass pestle in a homogenizer. The homogenate was centrifuged at 400 × g for 10 min at 4C. The pellet was recovered, washed twice in buffer N, and centrifuged for 10 min at 400 × g and 4C. The recovered nuclei were used fresh in buffer N or frozen at −80C in buffer N containing 70% glycerol.
An alternative method for isolating interphase nuclei was adapted from Suprynowicz and Gerace (1986). Harvested cells were washed in 40 volumes of isolation buffer (10 mM HEPES-KOH, pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 1 mM DTT, 10 μM cytochalasin B, 0.5 mM PMSF, 1 μg/ml pepstatin A, 1 μg/ml leupeptin, 1 μg/ml aprotinin), centrifuged for 6 min at 1500 × g, and resuspended in 3 volumes of isolation buffer and incubated on ice for 30 min. Swelled cells were homogenized with 50 strokes of a dounce homogenizer. The homogenate was layered over a cushion of 30% sucrose (w/v) in a buffer (10 mM HEPES-KOH, pH 7.4, 10 mM KCl, 1.5 mM MgCl2, and 1 mM DTT) and centrifuged for 2 min at 500 × g. Nuclei recovered in the pellet were treated as in the original method.
Digitonin-permeabilized cells were also used instead of a nuclear suspension. In a protocol adapted from nuclear import assays (Moore and Blobel 1992), unsynchronized HeLa cells grown on coverslips were permeabilized by a 5-min exposure on ice to 40 μg/ml digitonin in buffer A∗ (20 mM HEPES, pH 7.3, 110 mM potassium acetate, 5 mM sodium acetate, 2 mM magnesium acetate, 1 mM EGTA, 2 mM DTT, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatin) and washed in buffer A∗. Alternatively, in a protocol for CHOs by Wu et al. (1997), suspension cells recovered by exposure to trypsin/EDTA (as described above in the Cell Culture section) were used in place of isolated nuclei. Cells were recovered by centrifugation at 500 × g for 5 min, washed in ice-cold transport buffer (20 mM HEPES, pH 7.3, 110 mM potassium acetate, 2 mM magnesium acetate, 1 mM EGTA) at a concentration of 5 × 106 cells, centrifuged again, and resuspended in transport buffer at a concentration of 1 × 107 cells/ml. An equal volume of transport buffer containing 80 μg/ml digitonin was added, and the cells were incubated for 5 min on ice. An equal volume of 3% BSA in transport buffer was added to the mix to quench the remaining digitonin. The digitonin ghosts were recovered by centrifugation and resuspended in buffer N. These were treated as isolated nuclei from this point.
Permeabilization With Lysolecithin for Loading of Nuclei With Antibodies. According to the method of Collas et al. (1999), purified nuclei (2000/μl) were permeabilized in 500 μl of buffer N containing 0.75 μg/ml lysolecithin (Sigma) for 15 min at room temperature. Excess lysolecithin was quenched by adding 1 ml of 3% BSA in buffer N for 5 min on ice. Nuclei were sedimented at 400 × g for 10 min, washed once in buffer N, and sedimented again. Nuclei were resuspended in 100 μl of buffer N containing antibodies and incubated on ice for 1 hr with gentle agitation. Nuclei were sedimented at 500 × g through 1 M sucrose for 20 min and held in buffer N on ice until use.
Nuclear Disassembly. The following method was taken from Collas et al. (1999). A reaction consisted of 20 μl soluble mitotic extract, 1 μl nuclear suspension (∼2 × 104 nuclei), and 0.6 μl ATP-generating system (1 mM ATP, 10 mM creatine phosphate, and 25 μg/ml creatine kinase), which initiated the reaction. In the case of digitonin-permeabilized cells grown on coverslips, these were inversed on a drop of the incubation mixture (consisting of 20 μl soluble mitotic extract and 0.6 μl of the ATP-generating system described above) deposited on parafilm in a humid chamber. The reaction proceeded at 30C for up to 2 hr. Chromatin condensation was monitored by staining with 0.1 μg/ml Hoechst 33258.
Assembly Systems
Mitotic Homogenate for Nuclear Assembly. The procedure was adapted from Burke (1998). Mitotic cells were incubated in 100 volumes of complete medium containing 1 μM nocodazole and 20 μM cytochalasin B for 30 min at 37C. Cells were harvested by centrifugation at 1500 × g for 6 min and washed twice in 50 volumes of ice-cold PBS. Cells were washed in 1.5 ml of ice-cold KHM (78 mM KCl, 50 mM HEPES-KOH, pH 7, 4 mM MgCl2, 10 mM EGTA, 8.37 mM CaCl2, 1 mM DTT, 20 μM cytochalasin B) and centrifuged at 1000 × g for 5 min at 4C. Mitotic cells were resuspended in 1 volume of KHM and homogenized on ice with 10–25 strokes of a tight-fitting pestle in a 1-ml dounce homogenizer (aiming for 95% cell breakage).
Crude homogenate was used fresh by being diluted 2-fold with KHM and incubated for up to 2 hr at 37C to study nuclear reassembly. Alternatively, to obtain a postchromosomal supernatant containing mitotic membranes and cytoplasm, the homogenate was centrifuged at 1000 × g for 5 min at 4C to pellet chromatin, nuclei, and unbroken cells. This supernatant was used fresh or kept at −80C. In some experiments, 1 volume of postchromosomal supernatant was added to the crude homogenate before reassembly.
Digitonin System of Nuclear Assembly. This procedure was adapted from Kourmouli et al. (2000). Mitotic cells were quickly washed three times in ice-cold PIPES buffer (50 mM PIPES-KOH, pH 7.4, 50 mM KCl, 5 mM MgCl2, 2 mM EGTA, 1 mM PMSF), centrifuged for 30 sec at 1500 × g, and resuspended at a concentration of 106 cells/ml PIPES∗ buffer (PIPES buffer plus 1 mM DTT and 2 μg/ml each leupeptin, pepstatin A, aprotinin, and antipain). Cell membranes were permeabilized with 50 μg/ml digitonin (from a 1 mg/ml stock in DMSO) for 5 min on ice. Antibodies were sometimes added to 200-μl aliquots of the digitonin ghosts suspension, and the final volume was adjusted to 300 μl with PIPES∗ buffer. Suspensions were incubated at 33C for up to 2 hr.
Gel Electrophoresis and Immunoblotting
For total cell homogenates, cells were scraped from culture dishes in Tris-acetate buffer (10 mM Tris-acetate, pH 7.5, 150 mM NaCl, 1 mM EGTA) with a rubber policeman and transferred to conical centrifuge tubes. Cells were harvested by centrifugation at 1500 × g for 6 min and washed twice in the same buffer. For gel electrophoresis, cell pellets were resuspended in SDS-sample buffer to solubilize proteins and sonicated. Samples were boiled for 5 min before loading. Proteins were separated by electrophoresis in 5% stacking and 12% resolving SDS-polyacrylamide gels according to the method of Laemmli (1970).
Protein profiles were visualized by Coomassie blue [0.1% (w/v) Coomassie blue R-250, 10% acetic acid, 25% methanol] staining of gels. Otherwise, the separated polypeptides were electrophoretically transferred from gels to nitrocellulose membranes and processed for immunoblotting [Western blot (WB)] as recommended by Amersham Canada (Oakville, Ontario, Canada). Briefly, membranes were blocked for 1 hr in milk (5% low-fat powdered milk in PBS with 0.05% Tween) and incubated for 1 hr in primary antibodies. Detection was performed with the appropriate biotinylated secondary antibody followed by streptavidin-horseradish peroxidase (1:4000) and developed with a chemiluminescence kit (Amersham Canada). Reactivity was visualized on Hyperfilm ECL (Amersham Canada).
Indirect Immunofluorescence Staining
Intact interphase cells were fixed, permeabilized, and stained as previously described (Chaly et al. 1984). Briefly, cells attached to coverslips were washed in PBS twice for 30 sec and fixed for 5 min with 3% paraformaldehyde in PBS. They were washed again in PBS, reduced with 0.1% sodium borohydride (NaBH4) in PBS three times for 4 min, and permeabilized for 20 min in PBS containing 0.2% Triton X-100.
Cells were sometimes simultaneously fixed and permeabilized in ethanol. Mitotic cells in suspension were centrifuged 1 min at 700 × g to remove supernatant and resuspended in cold 95% ethanol and incubated at −20C for 15 min. Cells were centrifuged again to remove ethanol, resuspended in PBS, and kept on ice until all samples could be centrifuged on coverslips at 2000 × g for 10 min at 4C (plate rotor of a Hermle Z360K at 3500 rpm).
Mitotic cells in suspension, nuclei, or mitotic chromosomes were also centrifuged for 5 min at 1500 × g and resuspended in 3% paraformaldehyde in PBS for 5 min, centrifuged again, resuspended in PBS, and kept on ice until all samples could be centrifuged on coverslips at 2000 × g for 10 min at 4C (plate rotor of a Hermle Z360K at 3500 rpm). The material that adhered to coverslips was reduced with 0.1% sodium borohydride in PBS three times for 4 min and permeabilized for 20 min in 0.2% Triton X-100 in PBS.
Fixed and permeabilized material on coverslips was washed three times for 5 min in PBS, incubated for 1 hr with the primary antibody, washed again three times for 5 min in PBS, and incubated for 1 hr with the secondary antibody. This procedure was repeated if double immunolabeling was done. The cells were washed in PBS and stained with Hoechst 33258 (1 μg/ml in PBS), and coverslips were put cells side down on a drop of mounting medium (0.1% p-phenylenediamine and 50% glycerol in PBS). Conventional epifluorescence microscopy was carried out on a Zeiss Axiophot (Carl Zeiss Canada Ltd.; Toronto, Ontario, Canada), and images were captured with a Hamamatsu C5985 cooled CCD camera using Metamorph v 4.01 (Canberra-Packard; Ottawa, Ontario, Canada).
Antibodies
The primary antibodies used were the MAN antiserum at a dilution of 1:5000 previously described in Paulin-Levasseur et al. (1996); a mouse monoclonal antibody against lamin B1 (Oncogene; San Diego, CA) at a dilution of 1:50 for immunofluorescence (IF); a goat polyclonal antibody against the N terminus of lamins A/C (Santa Cruz) at a dilution of 1:100 for IF; a mouse monoclonal antibody against LAP2 termed 13d4 (provided by Dr. R. Benavente, University of Würzburg, Würzburg, Germany) at a dilution of 1:20 for IF, as previously described (Alsheimer et al. 1998); a mouse monoclonal antibody against α-tubulin termed DM1A (Sigma) at a dilution of 1:500 for IF; a goat polyclonal antibody against vimentin termed ZAK (provided by Dr. P. Traub, Max-Planck Institute for Cell Biology, Ladenburg, Germany) at a dilution of 1:1500 for IF and 1:3000 for WB; a mouse monoclonal antibody against nuclear pore complex proteins termed MAb414 (Babco Berkeley Antibody Company; Richmond, CA) at a dilution of 1:1500 for IF; a mouse monoclonal antibody against the 2H12 nucleolar protein at a dilution of 1:20 for IF; and a mouse monoclonal antibody against cyclin B1 (BD Biosciences Pharmingen; Franklin Lakes, NJ) at 1 μg/ml for WB.
The following secondary antibodies were used for IF: a rabbit anti-human IgG, IgA, and IgM conjugated with FITC (ICN ImmunoBiologicals; Lisle, IL) at a dilution of 1:400; a goat anti-human IgG conjugated with CY3 (Jackson ImmunoResearch Laboratories Inc.; West Grove, PA) at a dilution of 1:400; a rabbit anti-goat conjugated with FITC (MP Biomedicals Cappel; Irvine, CA) at a dilution of 1:250; and a donkey anti-mouse IgG conjugated with CY3 (Jackson ImmunoResearch) at a dilution of 1:400. Biotinylated secondary antibodies were used for WB: an anti-goat Ig and an anti-mouse Ig, both from Amersham and at a dilution 1:4000.
Results
Nuclear disassembly and assembly in mammalian cells are finely tuned processes involving the contribution of several proteins. The timely sequence of events leading to “deconstruction/reconstruction” of the nuclear compartment and the “cause/effect” of dynamic protein rearrangements during nuclear envelope fragmentation/reformation can be extremely difficult to establish in vivo, as shown by the double immunolocalization of LEM proteins and lamins A/C in control mitotic HeLa cells (Figure 1). Therefore, it seemed worthy to explore the possibility of using cell-free systems that could facilitate the dissection of these processes in mammalian cells through experimental manipulations.
Nuclear Disassembly in Cell-free Systems
The cell-free system tested at first for nuclear disassembly was based on the protocol developed for HeLa cells by the research team of Collas (Collas et al. 1999; Martins et al. 2000; Steen et al. 2000). With this system, interphase nuclei disassemble in the cyclin-containing soluble fraction of mitotic cytoplasm (a soluble mitotic extract isolated from nocodazole-synchronized cells) when supplemented with an ATP-generating system. The system can be manipulated by loading antibodies into the nuclei by lysolecithin permeabilization before exposure to the mitotic extract to block the action of specific proteins during disassembly.
Soluble mitotic extracts were obtained from HeLa cells by two different methods (see Materials and Methods for more details; extracts were isolated on three separate occasions for each method). On analysis by WB, both extracts were shown to contain cyclin B1 (Figures 2A and 2B, Lane 1) in the same proportion as whole mitotic cells by visual approximation (Figures 2A and 2B, Lane 2). Extracts were sometimes prepared by replacing EDTA with EGTA in the preparation buffer (see Materials and Methods), without any observed difference in results.
Nuclei were isolated also from HeLa cells, as described in Materials and Methods, but were consistently entangled in vimentin intermediate filaments that had collapsed onto the nuclear envelope during preparation (Figures 3A–3B″). These nuclei did not disassemble when exposed to mitotic extracts (Figures 3C and 3C′) but rather appeared to form clumps as incubation time elapsed (Figures 3D and 3D′). This was attempted numerous times with different batches of isolated nuclei. Analysis of the nuclei by immunoblotting further confirmed that the material surrounding them was at least partially composed of vimentin (data not shown).
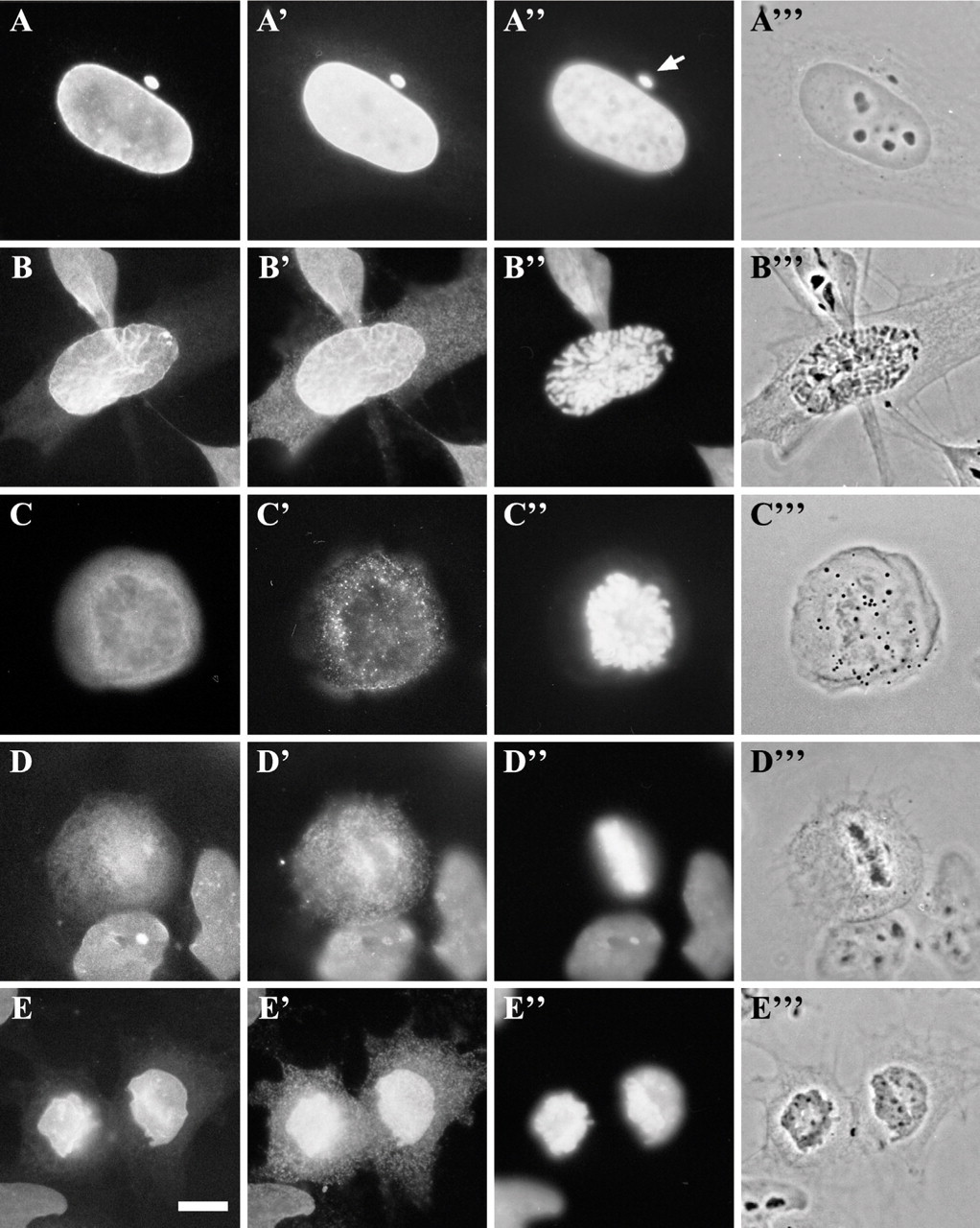
Spatial relationship of LEM proteins to lamins A/C throughout the cell cycle. Cells were double-labeled for immunofluorescence with the MAN antiserum (
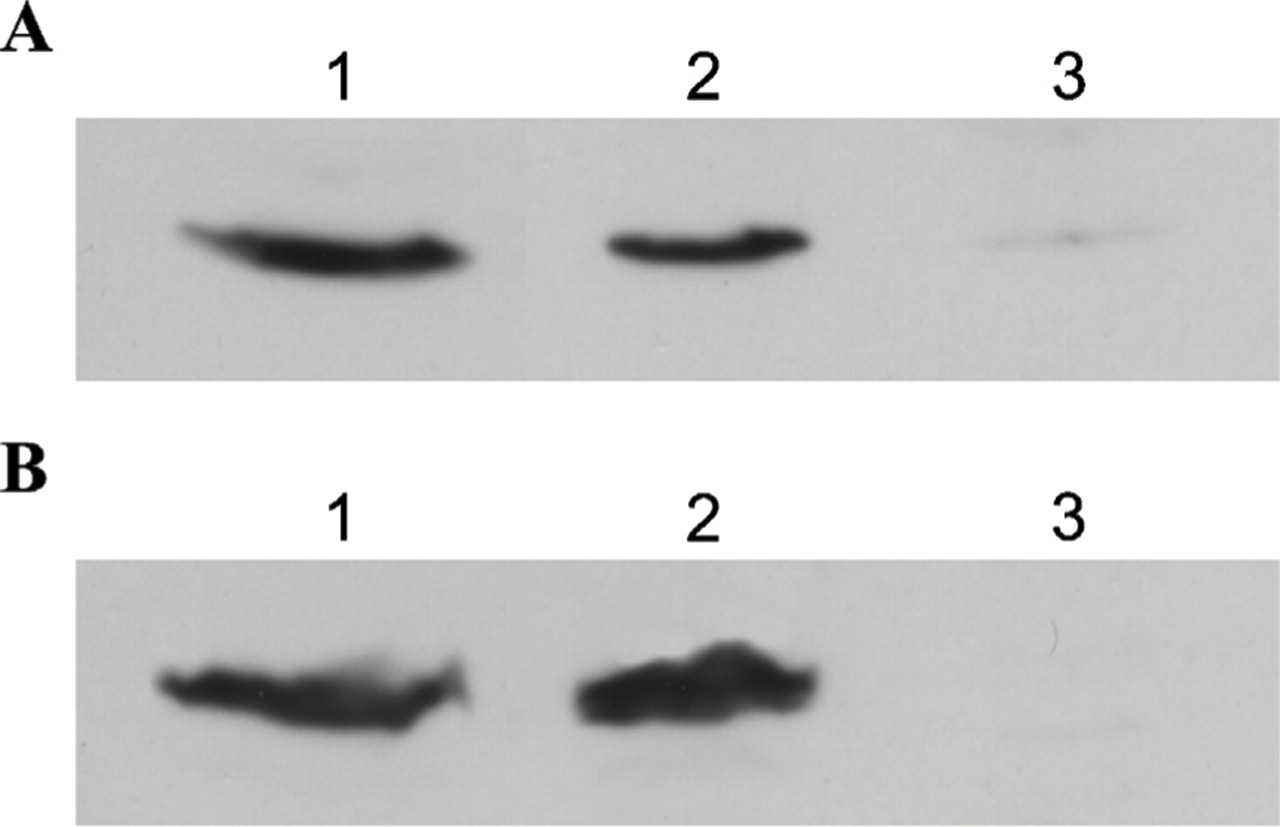
Cyclin B1 is present in mitotic soluble extracts. Proteins were separated by SDS-PAGE and immunoblotted with antibodies directed against cyclin B1. Mitotic HeLa cells were fractionated to obtain a mitotic soluble extract either by the method of Collas (
Some disassembly experiments were also performed with nuclei attached to coverslips beforehand (either by coating the coverslips with poly-L-lysine or by centrifuging the nuclei onto them) to reproduce in vivo conditions better, because HeLa cells grow attached to a substrate. However, the nuclei did not disassemble with the two different soluble mitotic extracts.
Furthermore, interphase nuclei isolated from CHO cells were used with the HeLa soluble mitotic extract. This was in the hope that, if the problem was with the HeLa nuclei alone, the cyclin-containing extracts would permit disassembly. Nevertheless, the CHO nuclei did not disassemble (results not shown). Many disassembly reactions were performed, with different batches of mitotic extracts and with nuclei isolated on different occasions, used fresh and thawed, but without success.
Because, in some experiments reported previously (Collas et al. 1999), the nuclei were permeabilized with lysolecithin to permit entry of antibodies before disassembly, the same treatment was applied to isolated nuclei before exposing them to mitotic extracts. Nuclei were indeed shown to be permeabilized by their incorporation of 2H12 antibodies after exposure to lysolecithin (Figures 3F and 3F′). However, lysolecithin-permeabilized nuclei did not disassemble more readily on addition of mitotic extracts on either of two attempts (Figures 3E and 3E′).
Because of the major difficulties encountered in obtaining a working nuclear disassembly system, other options were considered. Digitonin affects only the plasma membrane of cells and leaves the cytoplasm empty of most soluble components. This system is often used to study nuclear transport (Adam et al. 1990; Merle et al. 1999). It was hypothesized that the digitonin “ghosts” could be adapted to devise a novel nuclear disassembly assay. Thus, an asynchronous population of HeLa cells grown on coverslips was exposed on ice to digitonin. The coverslips were inverted on mitotic extracts containing an ATP-generating system and incubated at 30C for 2 hr in a humid chamber. The nuclei did not disassemble on all of three attempts. It was thought that attachment of digitonin ghosts to the substrate might prevent disassembly, because mitotic HeLa cells round up and are only loosely connected to the substrate when they are in culture. A suspension of interphase cells was therefore permeabilized with digitonin, rinsed, and exposed to mitotic soluble extracts. Again, the nuclei did not disassemble on both attempts.
Nuclear Assembly in Cell-free Systems
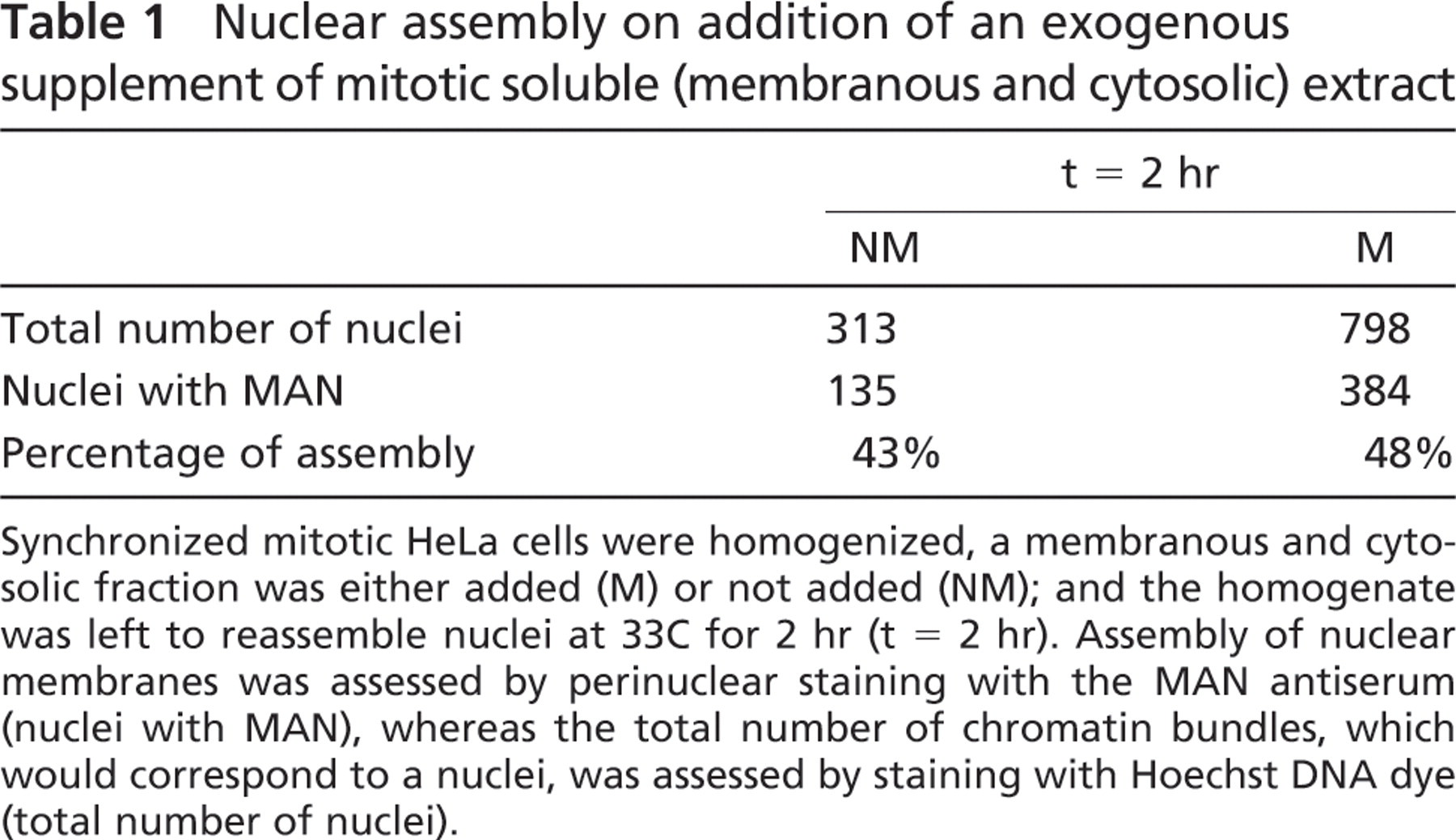
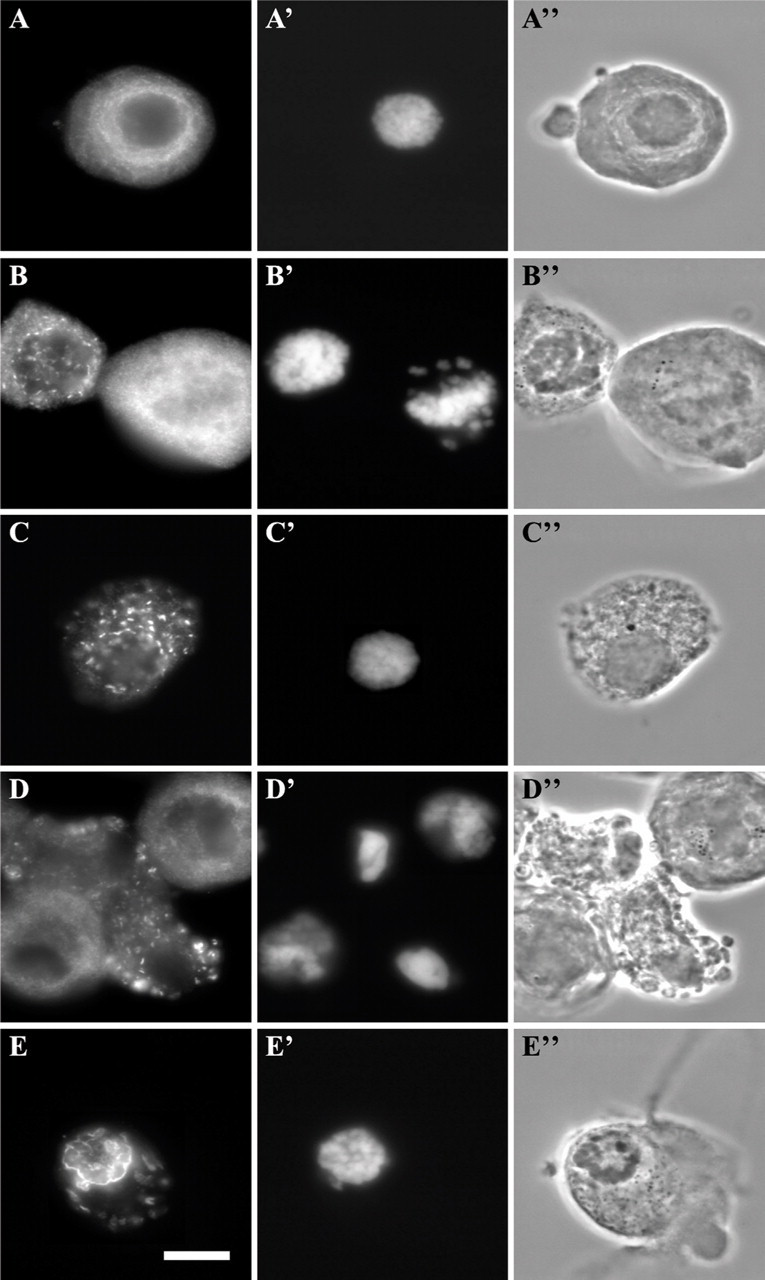
Incomplete Assembly in a Crude Mitotic Homogenate. The simplest method of cell-free nuclear assembly for mammalian cells is a crude homogenate of mitotic cells left to assemble nuclei in a buffer at 33C for up to 2 hr. A method developed by Burke and Gerace (1986) and Burke (1998) in CHO cells was adapted as the main protocol for HeLa cells. At time 0 (immediately after homogenization and before any incubation time), the condensed chromatin was devoid of marker nuclear antigens (Figures 4A–4A″), as was expected. After 2 hr of reassembly in the crude mitotic homogenate, the chromatin was found to be surrounded by incomplete nuclear envelopes as assessed by staining for different nuclear envelope antigens, including nucleoporins (Figures 4B–4B″), LAP2 proteins (Figures 4C– 4C″), LEM-bearing proteins (Figures 4D–4D″), and lamins A/C (Figures 4E–4E″). In some instances, chromatin masses presented a regular peripheral nuclear staining (see cells labeled with the MAN serum in Figure 4D), but this happened only when the nuclei were entangled with cytoplasmic material as visualized by phase contrast (Figure 4D″). A few nuclei that had apparently assembled without cytoplasmic material had also a relatively regular peripheral staining (Figures 4E–4E″), but these were extremely rare. In most cases, the staining of chromatin masses was not regular but rather discontinuous as shown in Figures 4C–4C″. Addition of a surplus of a postchromosomal supernatant containing mitotic membranes and cytoplasm to the mix (as suggested by Burke and Gerace 1986, described in Materials and Methods) improved the numbers of regularly stained nuclei only slightly (Table 1). In total, nine separate assembly reactions were attempted without satisfactory results.
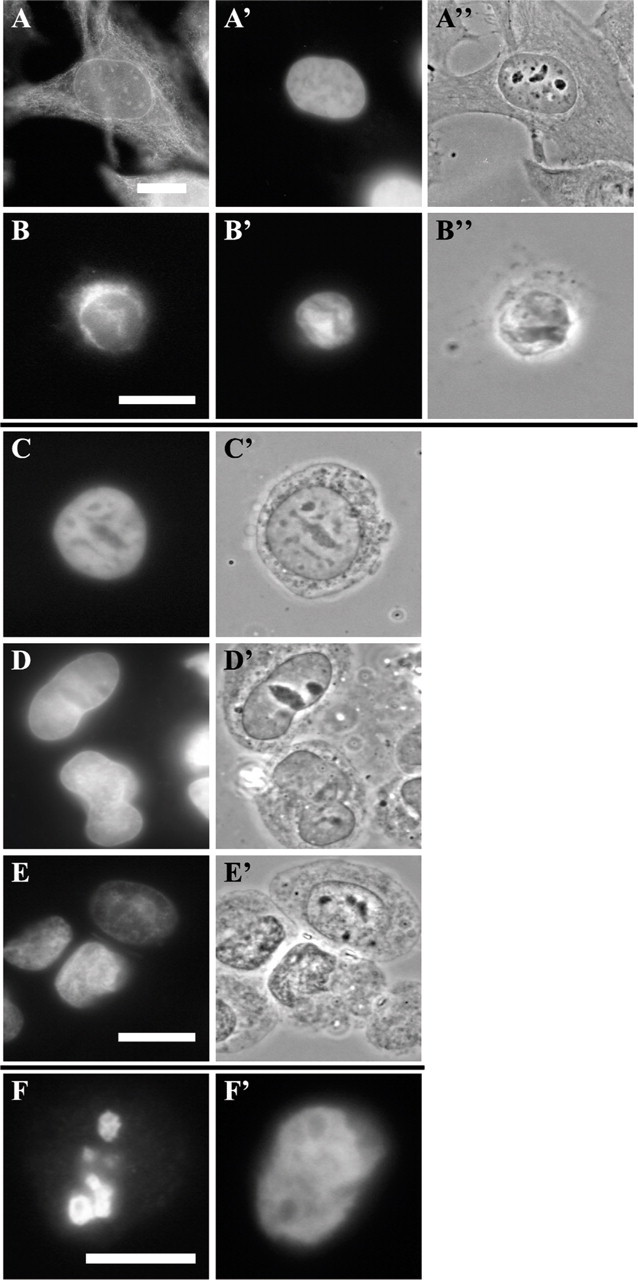
Isolated nuclei from whole HeLa cells failed to disassemble after 2 hr in mitotic soluble extract. Whole cells and isolated nuclei were labeled for immunofluorescence with anti-vimentin (
Incomplete assembly of nuclei in a crude homogenate of mitotic HeLa cells. Homogenate of mitotic cells at time 0 and after 2 hr at 37C. The material was labeled for immunofluorescence with anti-lamins A/C (
Incomplete Assembly in a Digitonin Cell-free System. As mentioned, digitonin was used in an unsuccessful attempt at nuclear disassembly. However, a digitonin system had been used by Kourmouli et al. (2000) to test the effects of recombinant forms of HP1 on nuclear envelope assembly. In the protocol elaborated by this group, synchronized mitotic cells are exposed to digitonin for permeabilizing the plasma membrane and allowing the entry of peptides or exogenous proteins, and left at 33C for 2 hr for the nuclei to reassemble (see Materials and Methods for details). Assembly reactions were attemptedon10separate occasions. Microscopic analysis of mitotic cells immediately after permeabilization indicated a diffuse cellular localization of marker nuclear antigens (Figures 5A–5A″ and 5D–5D″). After 30 min of incubation, LAP2 proteins and lamin B formed cytoplasmic inclusions (Figures 5B–5B″ and 5E–5E″). After 2 hr, the assembly of nuclear envelopes was still incomplete in most cases as assessed by staining for LAP2 proteins (Figures 5C–5C″) and lamin B (Figures 5F–5F″). Cytoplasmic aggregates of antigens were again present. Also, the chromatin remained condensed, and the nuclei did not grow in size (Figures 5C′ and 5F′), even after 4 hr of incubation (results not shown). Similar results were obtained with CHO cells (results not shown).
Nuclear assembly on addition of an exogenous supplement of mitotic soluble (membranous and cytosolic) extract
Synchronized mitotic HeLa cells were homogenized, a membranous and cytosolic fraction was either added (M) or not added (NM); and the homogenate was left to reassemble nuclei at 33C for 2 hr (t = 2 hr). Assembly of nuclear membranes was assessed by perinuclear staining with the MAN antiserum (nuclei with MAN), whereas the total number of chromatin bundles, which would correspond to a nuclei, was assessed by staining with Hoechst DNA dye (total number of nuclei).
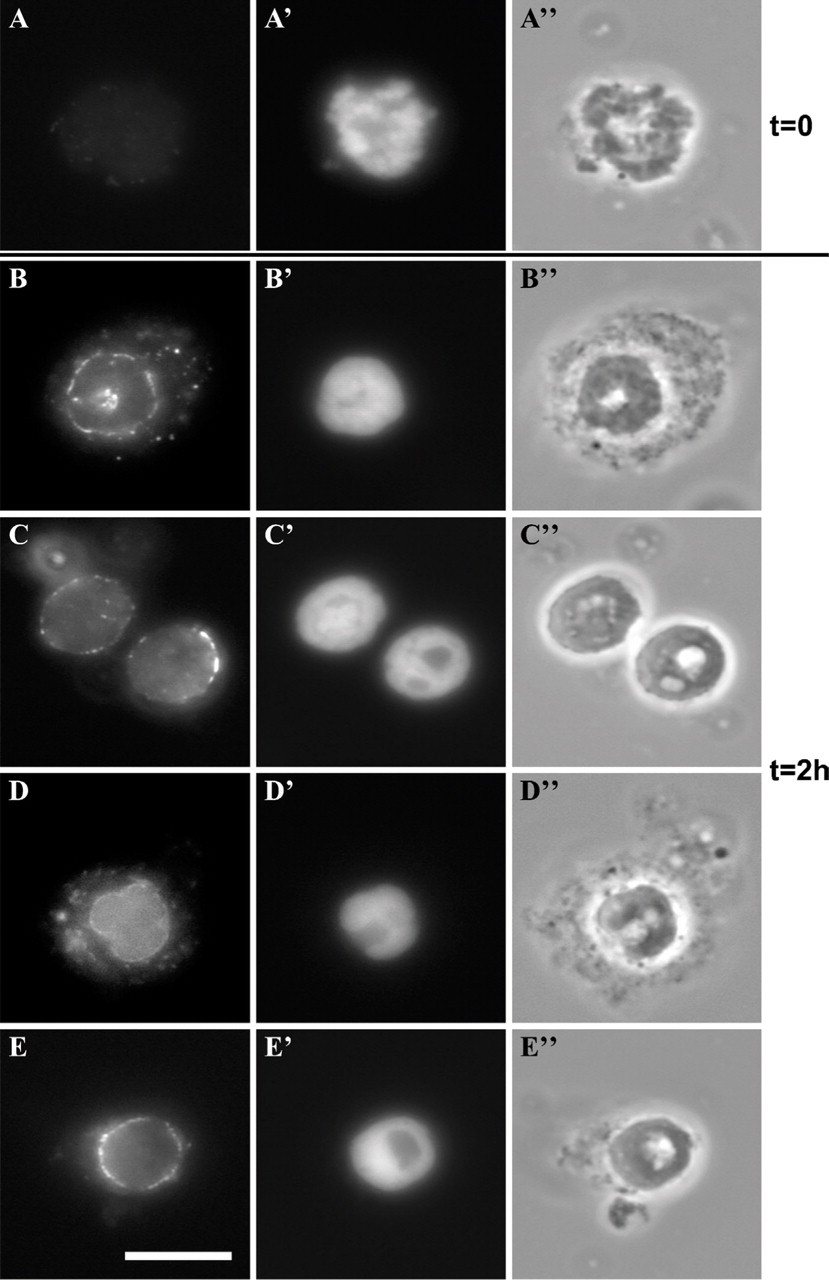
Poor Yield of In Vivo Nuclear Assembly Among Nocodazole-synchronized Cells. After the poor results obtained with the methods described above, the in vivo assembly of nuclei in nocodazole-synchronized cells was assessed. HeLa cells were released from an 18-hr nocodazole block and incubated at 33C for up to 2 hr, either in the buffer used for the assembly experiments with digitonin-permeabilized cells or in complete culture medium. Immediately after nocodazole block release, LAP2 antigens were in soluble or membrane-bound form in the cytoplasm (Figures 6A–6A″). Surprisingly, a relatively high number of inter-phase cells could be seen at the start of the experiment. These cells, representing in average 18% of the populations (Table 2), had thus escaped synchrony. At 10 min after the release, LAP2 proteins started to associate with chromatin (Figures 6B–6B″). After 20 min, these proteins were found in cytoplasmic inclusions and started to assemble around the chromatin in some cells (Figures 6C–C″). The yield of complete assembly was very low after 2 hr of incubation (Figures 6D–6D″ and 6E–6E″; Table 2), and the chromatin did not seem to decondense (Figures 6D′ and 6E′).
Microtubule organization was used also to evaluate nuclear assembly in intact versus digitonin-permeabilized nocodazole-synchronized cells. At the start of the release, the tubulin was in soluble form in the cytoplasm of both intact and permeabilized cells (Figures 7A–7A″ and 7B–7B″). After 45 min of incubation, the majority of intact nocodazole-synchronized cells had assembled an aberrant number of mitotic spindle poles, e.g., three or more (Figures 7C–7C″, cell on the right), compared with the normal bipolar spindle (Figures 7C–7C″, cell on the left) of unsynchronized cells. Comparatively, permeabilized cells had lost most of their tubulin and displayed no staining (Figures 7D–7D″).
Discussion
Cell Synchrony
Unsynchronized wild HeLa populations, like most cultured mammalian cell lines, include a limited number of cells in mitosis, namely a proportion of ∼5%. To obtain sufficient numbers of mitotic figures for experimentation, cells can be synchronized by M-phase block, for instance, by reversibly disrupting microtubule functions. In this study, HeLa cells were exposed to nocodazole, which arrests cells in prometaphase by inhibiting assembly of the mitotic spindle necessary for chromosome separation (Merrill 1998). The cells were further isolated by mechanical shake-off (Terasima and Tolmach 1963; Tobey et al. 1967), which selectively detaches rounded M-phase cells. Mammalian cells treated with anti-microtubule drugs progress through interphase but are arrested at mitosis. According to the literature, the human HeLa cell line completes mitosis after release from a nocodazole arrest (4 hr at 0.04 μg/ml or 0.13 μM, with a preliminary thymidine block) in an average of 80 min, with >80% of the spindles appearing to be bipolar (Zieve et al. 1980). If cells are arrested for extended periods in mitosis or with higher concentrations of drug, mitotic spindles reform less efficiently with a large percentage of cells incapable of returning to interphase (Zieve et al. 1980). Prolonged exposures to nocodazole also cause irreversible polyploidization and the formation of micronuclei when some cells exit mitosis without dividing (Miller-Faurè s et al. 1981; Nüsse and Egner 1984). Hence, short exposures to small concentrations of nocodazole would seem in theory to be the optimal way, but it can provide only small numbers of mitotic cells.
For the experiments described in this study, a conservative exposure to 1 μM nocodazole for 18 hr was used, as in many reports (Collas et al. 1999). After the cells were released, aberrant numbers of mitotic spindles (three or more) were observed in more than one half of the cells. Furthermore, 60% of cells released from a nocodazole block and cultured in complete medium failed to attach subsequently to the substrate (data not shown). It was also previously reported that a 24-hr nocodazole block (0.1 μM) of a human malignant glioma cell line resulted in death for the majority of the cells after 24 hr of recovery (Hueber et al. 1998). Presumably, nocodazole block has detrimental effects. Nonetheless, Klein et al. (1997) found that nocodazole-synchronized (2.5 μM for 24 hr) HeLa cells had a 95% viability rate by dye exclusion. Likewise, nocodazole has been used in many studies where the results obtained from synchronized and unsynchronized cells are comparable. For example, up-regulated activity of LIM-kinase 1 was monitored in mitotic cells and prometaphase-synchronized cells (Sumi et al. 2002). Nucleoplasmic coiled bodies were also analyzed for expression of p80-coilin in cell populations synchronized by many methods, including nocodazole block, and the results were similar (Andrade et al. 1993).
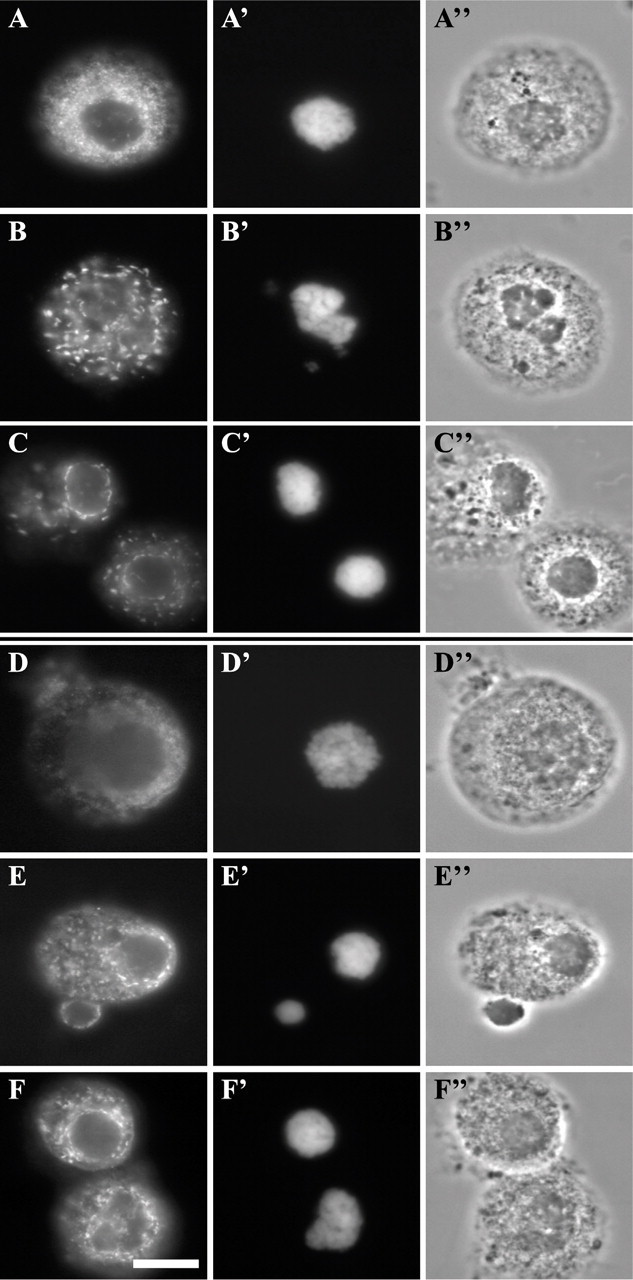
Incomplete assembly of nuclear markers in nuclei of digitonin-permeabilized mitotic HeLa cells. Cells were labeled for immunofluorescence with anti-LAP2 proteins (
Incomplete assembly of LAP2 proteins in nuclei of nocodazole-synchronized HeLa cells. Cells were labeled for immunofluorescence with anti-LAP2 (
Nuclear assembly of LAP2 in synchronized HeLa cells
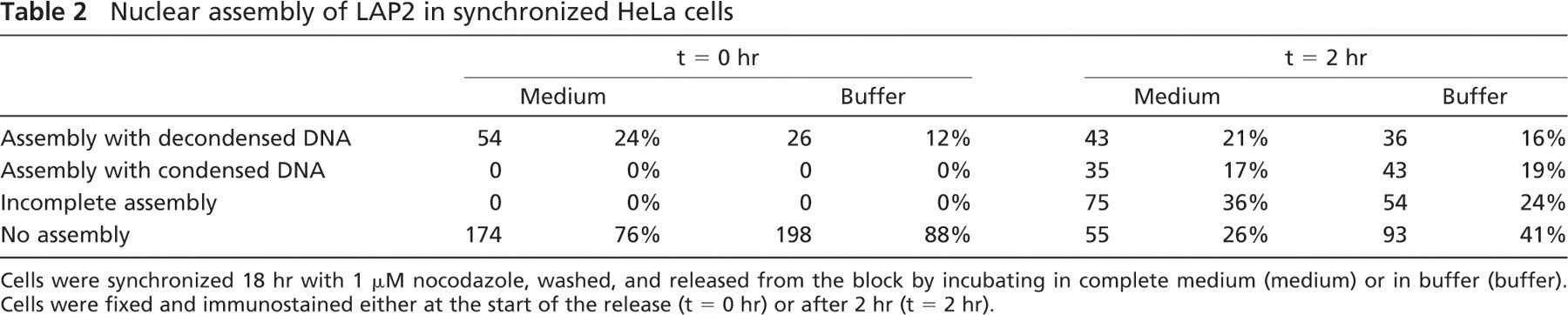
Cells were synchronized 18 hr with 1 μM nocodazole, washed, and released from the block by incubating in complete medium (medium) or in buffer (buffer). Cells were fixed and immunostained either at the start of the release (t = 0 hr) or after 2 hr (t = 2 hr).
Although the nocodazole “batch” synchronization was necessary in the case of our study to get enough mitotic cells, this method suffers many drawbacks. It has been argued that even though batch treatments are extensively used and traditionally considered valid, they simply do not result in a synchronized cell population (Cooper 1998, 2003). For instance, induced G0 arrest, or G1 phase arrest, by starvation or inhibition, may be the most used approach to cell cycle analysis. The traditional way of defining a G1 cell block is a 2n DNA content. Cells are considered to resume cell cycle synchronously when they are released from the block. However, because growth was inhibited, they have a wide size distribution. Because they need to get to a normal size before entering S phase, the resulting cell population is theoretically no more synchronized than before (Cooper 1998). It has also been shown that nocodazole treatment does not result in a truly synchronized cell population (Cooper et al. 2006). Relative cell size distribution of nocodazole-treated cells was not narrowed, and when released, these cells did not proceed normally through the cell cycle, pointing to a deleterious or damaging effect of the drug. This suggests that the rapid loss of synchrony encountered in induction methods could be because of the fact that they are, in reality, not synchronized.
Induction methods can, however, still be used to examine a particular cellular feature, for example, condensed chromatin lacking a nuclear envelope in the case of nocodazole block (Cooper 2002). However, even then, meaningfulness of the observations can be arguable. The method of synchronization and the cell line can engender different results. This is the case for the cell-cycle effect of DNA-end binding (DEB) activity of Ku, the DNA-binding component of the protein kinase, DNA-PK. Although DEB activity varied during the cell cycle of chemically synchronized cells, cells from an unsynchronized population separated by centrifugal elutriation into distinct mitotic phases showed no difference in DEB activity (Chou and Chou 1999). Moreover, some have observed that “chemically” treated cells show growth imbalance and higher variation of all measured parameters, including cyclin expression and the degree of pRB phosphorylation (Gong et al. 1995).
In this study, metaphase nocodazole synchronization of HeLa cells resulted in incomplete and aberrant assembly of nuclei compared with an unsynchronized population. Nuclei assembled for 2 hr from nocodazole-synchronized cells showed incomplete nuclear envelopes and condensed chromatin. Also, assembly did not appear to proceed in the same way as in unsynchronized cells. During nuclear assembly of nocodazole-synchronized cells, cytoplasmic inclusions of INM antigens were observed. Similar inclusions could be found in control cells at the end of telophase but were much more prevalent in cells released from a nocodazole block than in unsynchronized HeLa.
Validity of Cell-free Systems of Nuclear Disassembly and Assembly
In this study, it was observed that the HeLa and CHO cell-free systems of nuclear disassembly and assembly give unreliable results. Nuclear disassembly was attempted numerous times with different batches of mitotic extracts and isolated interphase nuclei, but without success. Immunofluorescence microscopy showed that the material surrounding isolated nuclei contained vimentin, a cytoplasmic intermediate filament subunit. It is unclear how vimentin may possibly interfere with disassembly, because intermediate filaments are normally present in cells and do not necessarily disassemble during cell division. During mitosis, vimentin is known to form a “cage” around prometaphase chromosomes and the mitotic spindle (Aubin et al. 1980; Maison et al. 1993). The presence of vimentin around isolated nuclei could therefore hardly account for the lack of disassembly.
It is generally thought that the phosphorylation of INM and chromatin proteins may be the only necessary step in completing nuclear disassembly (reviewed in Margalit et al. 2005). Nevertheless, the mitotic extracts to which isolated nuclei were exposed in this study were shown to contain cyclin B1 (Figure 1) and should have supported nuclear disassembly. However, these nuclei had been isolated from unsynchronized cells and, therefore, displayed heterogeneity in their DNA and protein content. In the context of a mammalian cell-free system where the nuclear disassembly machinery is most likely synthesized/activated at strategic time points during the cell cycle, such a heterogeneity might have been a major limitation to efficient batch nuclear disassembly.
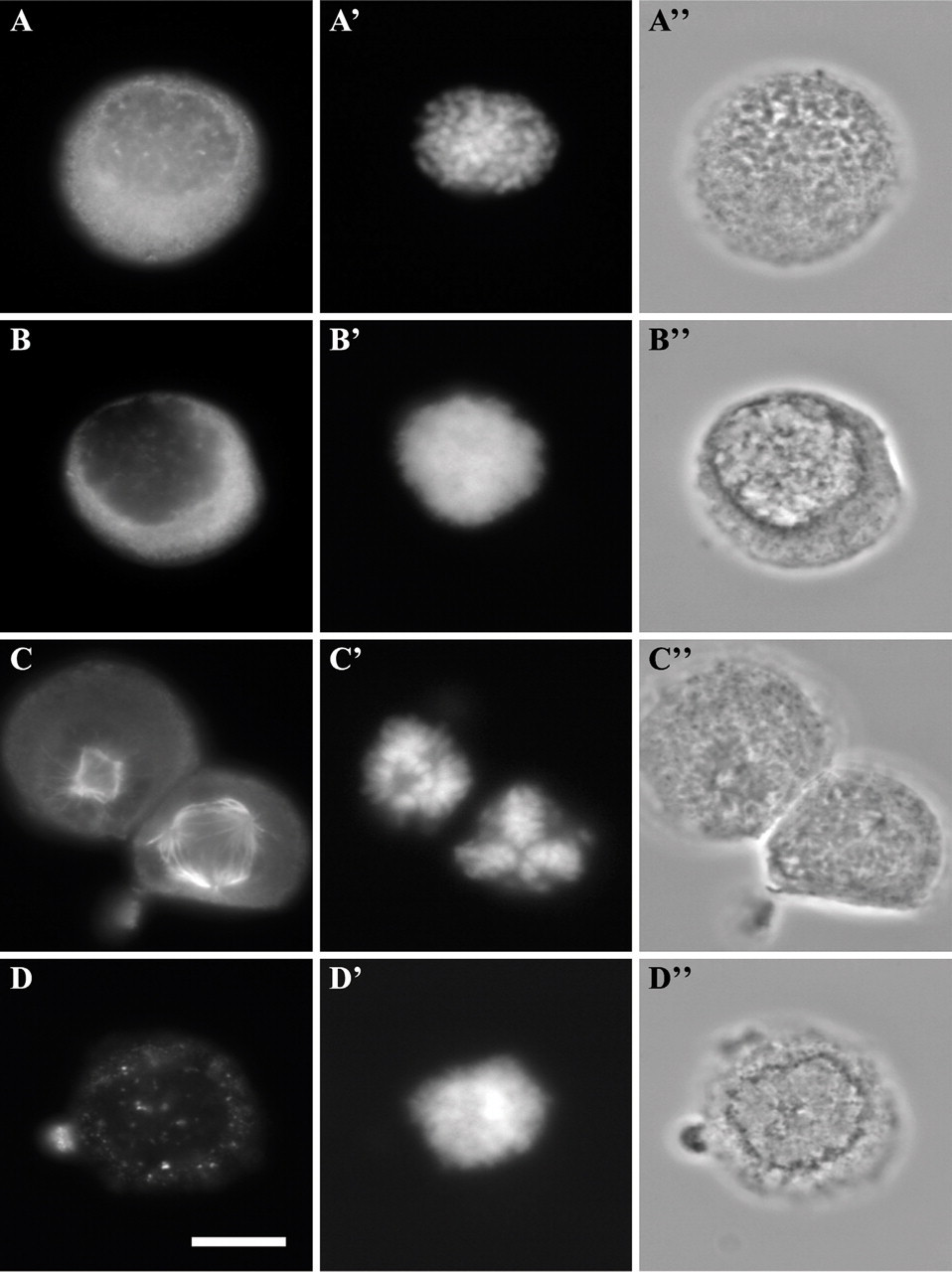
Tubulin distribution in intact or digitonin-permeabilized, nocodazole-synchronized HeLa cells. Cells were labeled for immunofluorescence with anti-tubulin (
The present study has also shown that the mitotic synchronization of HeLa cells by nocodazole block results upon subsequent release in incomplete and abnormal nuclear assembly of INM proteins such as the LEM proteins, including LAP2 polypeptides. The mechanism behind the nocodazole block is that mitotic spindles will not form, halting mitosis at prometaphase and preventing subsequent steps of the cell cycle. Because it has become clear that the mitotic spindle plays an important part in disassembly by pulling the NE apart in a specific manner (Beaudouin et al. 2002; Salina et al. 2002), NEBD without mitotic spindles, as it happens in nocodazole-blocked cells, cannot proceed as in vivo. Presumably, an aberrant disassembly will not result in a “normal” reassembly as cells are released from the block. Furthermore, the abnormal number (three or more compared with two) of mitotic spindles could affect chromosome segregation, consequently altering assembly. If the block itself affects assembly, inferences made from nocodazole-synchronized cells will not reflect in vivo events.
In cell-free systems of assembly (homogenates and digitonin-permeabilized mitotic cells), mitotic spindles are totally absent. They are also lacking in cell-free systems of nuclear disassembly, theoretically leading to a NEBD based solely on the weakening associations of nuclear components caused by hyperphosphorylation and not on the specific pattern of tearing initiated by astral microtubules. Because many studies have used cell-free systems of nuclear disassembly and assembly, their relevance to in vivo conditions needs to be reexamined.
Although the many cell-free systems have laid the groundwork for a biochemical dissection of nuclear disassembly and reassembly, evidence from cell-free systems is sometimes contradicted by observations made in whole cells. An in vivo electron-spectroscopic imaging study of fixed HeLa seemed to show that nuclear envelope cisternae preassemble in the cytoplasm before docking to the chromatin and gradually forming mature envelopes (Stracke and Martin 1991). Evidence for the coating of newly segregated chromatids with ER-like cisternae also comes from live cells (Ellenberg et al. 1997). Conflicting with these observations, NE assembly in cell-free systems occurs at the surface of chromosomes (Burke and Gerace 1986; Collas et al. 1996; Lopez-Soler et al. 2001). However, our experiments with intact (Figure 6) and digitonin-permeabilized (Figure 4) synchronized mitotic cells showed cytoplasmic inclusions of inner nuclear membrane proteins in great numbers as assembly progresses. These might be nuclear envelope cisternae preforming in the cytoplasm, making the digitonin system closer to in vivo conditions than cell-free systems based on homogenates.
Despite their drawbacks, cell-free systems remain an invaluable tool for the study of nuclear breakdown and assembly because they are still the only way to isolate these processes for study. However, we should not assume results obtained from cell-free studies will all apply to live cells. This study even leads us to believe that cell-free systems of nuclear assembly using nocodazole as a cell-cycle synchronizing agent should not be used, as all results may be artifactual from the effects of the drug. In this case, it may be useful to rely more on in vivo studies for future research. However, if cell-free systems are here to stay, the effects of cell synchronization, particularly nocodazole block, on nuclear assembly need to be thoroughly revisited.
Footnotes
Acknowledgements
This work was supported by a grant from the Natural Sciences and Engineering Research Council of Canada.