
Abstract
During germinal center (GC) reactions, B-lymphocytes with high-affinity B-cell receptors are selected. Regulation of apoptosis is a key process in selecting such wanted B-cells and in eliminating B-cells with unwanted specificities. In this paper, we show that apoptosis in human GC B-cells involves lysosomal destabilization, which is strictly controlled by caspase-8 activity, but not by caspase-9 activity. Ligation of CD40 provides resistance to lysosomal destabilization. Experimental lysosomal rupture by the lysosomotropic drug O-methyl-L-serine dodecylamide hydrochloride (MSDH) induces apoptosis in GC B-cells, including phosphatidyl serine exposure, mitochondrial inactivation, and DNA fragmentation. These apoptotic features occur in the absence of caspase-3 activity. Follicular dendritic cells (FDCs) protect binding B-lymphocytes from lysosomal destabilization, in both the absence and the presence of MSDH. Our study demonstrates that lysosomal leakage induces apoptosis of GC B-cells in a caspase-3-independent manner and that high-affinity binding to FDCs prevents lysosomal leakage and apoptosis in GC B-cells.
Keywords
G
FDCs present native antigens as immune complexes to B-cells in GCs. Due to competition, only B-cells with the highest affinity BCRs bind to these antigens and receive survival signals from the FDCs (Lindhout et al. 1993; Van Eijk et al. 2001a). Low-affinity, non-binding B-cells rapidly die by apoptosis.
The mechanisms by which FDCs silence apoptosis in GC B-cells have been clarified only partially. Apoptosis in GC B-cells is mainly induced via the death receptor pathway (Hennino et al. 2001; Van Eijk et al. 2001a,b). In freshly isolated GC B-lymphocytes, Fas (CD95), procaspase-8, Fas-associated death domain-containing protein (FADD), and the long isoform of cellular FLIP (cFLIPL) are included in an inactive death-inducing signaling complex (DISC) (Hennino et al. 2001). Upon culture of isolated cells at 37C, cFLIPL rapidly dissociates from the DISC, and this results in activation of caspase-8 (Hennino et al. 2001). Maintenance of cFLIPL association with the DISC is essential to prevent caspase-8 activation and consequent apoptosis. cFLIPL association with the DISC can be maintained by ligation of either the BCR or CD40, but also by signals provided by FDCs (Hennino et al. 2001; Van Eijk et al. 2001a,b,2003). The nature of the FDC-derived signals that prevent cFLIPL dissociation in adhering B-cells is poorly understood, but they act independently of signaling through CD40, BCRs, or adhesion receptors (Bonnefoy et al. 1993; Koopman et al. 1991,1994,1997; Lindhout et al. 1995; Van Eijk et al. 2001b,2003).
We have previously shown that apoptotic DNA fragmentation in GC B-lymphocytes can be blocked not only by inhibition of caspase-8 or caspase-3, but also by inhibition of cathepsins (Van Eijk and De Groot 1999; Van Eijk et al. 2003). Because cathepsins are mainly lysosomal enzymes, we decided to investigate in more detail a possible role of lysosomes in GC B-cell apoptosis. Lysosomal destabilization has been implied in apoptosis under the influence of several stimuli, including lysosomotropic drugs (Li et al. 2000; Kagedal et al. 2001; Boya et al. 2003a,b), oxidative stress (Brunk and Svensson 1999; Zhao et al. 2001,2003), serum withdrawal (Brunk and Svensson 1999), tumor necrosis factor (Guicciardi et al. 2000; Foghsgaard et al. 2001), and triggering of Fas (Brunk and Svensson 1999). As a result, lysosomal proteases such as cathepsins translocate from the lysosomes to the cytosol. It has been shown that several cathepsins, especially the cysteine cathepsins B and L and the aspartyl cathepsin D, participate in apoptosis in a caspase-dependent (Guicciardi et al. 2000; Kagedal et al. 2001; Cirman et al. 2004) and a caspase-independent manner (Biggs et al. 2001; Foghsgaard et al. 2001; Broker et al. 2004). One of the possible mechanisms through which cathepsins trigger apoptosis is cleavage of the Bcl-2 family member Bid (Stoka et al. 2001; Reiners et al. 2002; Cirman et al. 2004), resulting in mitochondrial inactivation.
We applied the lysosomotropic drug O-methyl-L-serine dodecylamide hydrochloride (MSDH), which specifically induces lysosomal destabilization (Li et al. 2000; Boya et al. 2003b) to GC B-cells. MSDH accumulates in the lysosomes by protonation and induces lysosomal leakage and translocation of cathepsins into the cytosol, resulting in apoptotic features (Wang et al. 1998; Li et al. 2000; Boya et al. 2003b).
Here, we demonstrate that lysosomal destabilization in GC B-lymphocytes is an early event in apoptosis and is under the control of caspase-8 activity. Induction of lysosomal leakage in GC B-cells results in cathepsin B activity outside lysosomes, and apoptotic features, including phosphatidyl serine (PS) exposure, mitochondrial inactivation, and the formation of DNA strand breaks, in the absence of caspase-3 activation.
Materials and Methods
Isolation of GC B-cells
B-lymphocytes were isolated from tonsils obtained from children undergoing routine tonsillectomy, as described previously (Lindhout et al. 1995). Briefly, tonsillar cell suspensions were depleted of T-cells using 2-(2-aminoethyl)-isothiourea dihydrobromide-treated (Sigma-Aldrich Chemie; Zwijndrecht, The Netherlands) sheep red blood cells, followed by density centrifugation on Lymphoprep (1.077 g/ml; Axis-Shield PoC AS, Oslo, Norway). The resulting cell population usually contained >95% CD20+ cells (B-cells) and <5% CD3+ cells (T-cells). This B-cell suspension was incubated with mouse monoclonal antibodies (MAbs) against IgD (Oxford Biotechnology Limited; Oxfordshire, UK) and CD39 (Ancell Corporation; Bayport, MN). Labeled cells were depleted using sheep anti-mouse Ig-coated Dynabeads (Dynal ASA; Oslo, Norway). This procedure normally yielded >95% pure CD38+IgD- GC B-cells.
Isolation of FDC-enriched Fractions
FDCs were isolated from tonsils as described by Parmentier et al. (1991). Briefly, tonsils were cut into pieces and treated with Iscove's modified Dulbecco's medium (IMDM)(Cambrex Bio Science Verviers; Verviers, Belgium) plus 200 U/ml collagenase type IV (Worthington Biochemical Corporation; Lakewood, NJ) and 7 U/ml DNase I (Roche Diagnostics; Mannheim, Germany), followed by density sedimentation on an ice-cold discontinuous gradient consisting of layers of 2.5% and 5% Path-O-Cyte 4 (MB-Biomedicals; Irvine, CA) in IMDM. The FDC-enriched fraction at the 2.5-5% interface was harvested, washed twice, and used for further experiments.
Cell Cultures
All cell cultures were made in IMDM supplemented with 86 mg/l gentamycin (Duchefa; Haarlem, The Netherlands) and 10% fetal calf serum (FCS) (Hyclone; Logan, UT) in 6- or 24-well plates (Corning Inc; Corning, NY). GC B-cells were seeded at a density of 2.106 cells/ml and cultured at 37C for indicated time periods in the presence or absence of several agents: α-CD40 MAb (at indicated concentrations; kind gift of Dr. M. de Boer, Pangenetics; Utrecht, The Netherlands), 100 μM z-IETD-fmk (IETD) or Ac-LEHD-cmk (LEHD) (both from Bachem AG; Bubendorf, Switzerland), 100 μM z-VAD-fmk (ZVAD; Alexis, Breda, The Netherlands), and 20 μM MSDH (kind gift of Dr. Gene Dubowchik, Pharmaceutical Research Institute, Bristol-Myers Squibb; Wallingford, CT). IETD, ZVAD, LEHD, and MSDH were dissolved in DMSO. α-CD40 MAb was dissolved in IMDM.
FDC-enriched cell suspensions were cultured in 75-cm2 culture flasks at 107 cells/ml for 16 hr at 37C. Next, FDC/B-cell clusters were separated from single B-cells by sedimentation at 1 × g on IMDM with 30% FCS, and clusters were isolated from the pellet. For detachment of B-cells from FDCs, clusters were incubated with 1.2 mM EDTA in PBS for 15 min at 37C. Single B-cells were analyzed for lysosomal destabilization and apoptotic parameters as described below. For confocal scanning laser microscopy (CSLM), FDC/B-cell clusters were plated in a 6-well culture plate on cover glass (18 × 18 mm) and after 16 hr incubation at 37C, clusters were stained as described below.
Lysosomal Integrity Assays
Cells were assessed for lysosomal stability at different time points in three different ways. Live B-cells were stained with 0.5 μg/ml acridine orange (AO; Sigma-Aldrich Chemie) or 100 nM LysoTracker Green (LTG; Molecular Probes, Eugene, OR) (Parmentier et al. 1991; Zhao et al. 2001) for 30 min on ice. After washing with PBS, cells were analyzed by CSLM, recording green (for LTG) or red (for AO) fluorescence, or by flow cytometry using a FACScan (Becton-Dickinson; Mountain View, CA).
Localization of Cathepsin B Activity in Live B-cells
Activity of cathepsin B was localized in live B-cells as a marker of lysosomal content at different time points using Z-Arg-Arg-cresyl violet as selective fluorogenic cathepsin B substrate (Van Noorden et al. 1998; Boonacker and Van Noorden 2001). Incubations were performed at 25C on the stage of a DM RA HC fluorescence microscope (Leica; Wetzlar, Germany) with a filter cube containing a custom-made excitation filter of 570-590 nm and an emission filter of 600-660 nm (Chroma; Rockingham, VT) to specifically localize formation of cresyl violet in B-cells as a cleavage product of cathepsin B activity (Boonacker et al. 2003). Formation of fluorescence of cresyl violet was followed in time and images were taken using a PLAN APO 100X oil-immersion objective and a cooled charge-coupled device camera (KX 1400; Apogee Instruments, Carlsbad, CA) at 2-5 min after cells and incubation medium [2 μM [Z-Arg-Arg]2-cresyl violet (Enzyme Systems Products and Prototek; Livermore, CA) in PBS, pH 7.4] were mixed on a glass slide and covered with a coverslip.
Before the cytochemical detection of cathepsin B activity, B-cells were either not cultured (freshly isolated B-cells) or cultured for 16 hr in the presence or absence of 100 μg/ml α-CD40 MAb, 100 μM IETD, 100 μM LEHD, or 100 μM ZVAD.
Detection of Apoptotic Parameters
Reduction of mitochondrial membrane potential was analyzed with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1; Molecular Probes) as described previously (Salvioli et al. 1997; Yuan et al. 2002). Stock JC-1 was made in DMSO and applied to the cells at concentrations of 5 μg/ml for 15 min at 37C. Cells were washed in PBS, and the shift from red to green fluorescence was measured by flow cytometry.
DNA strand breaks were analyzed using the In Situ Cell Death Detection Kit (Roche Diagnostics) according to the instructions of the manufacturer.
PS exposure was determined using Annexin-V-FITC conjugate (BioSource International; Camarillo, CA) in combination with propidium iodide (BioSource). Cells were stained according to the manufacturer's instructions and analyzed on a FACScan. For CSLM, cells were stained with an Annexin V-biotin kit (Immunotech; Marseille, France) according to the manufacturer's instructions, followed by incubation with streptavidin-Cy3 (Sigma-Aldrich Chemie).
Caspase-3 activity was estimated using a phycoerythrin-labeled MAb recognizing active caspase-3 (Pharmingen; San Diego, CA). One hundred thousand cells were fixed with 4% formaldehyde (Sigma-Aldrich Chemie), permeabilized with 0.5% saponin (Sigma-Aldrich Chemie), and incubated for 30 min on ice with 10 μl of the α-caspase-3 antibody. Cells were washed with PBS and analyzed by flow cytometry.
CSLM
FDC/B-cell clusters cultured on coverslips and single GC B-cell suspensions were examined using an SP2 CSLM (Leica Microsystems; Rijswijk, The Netherlands). A X63 oil-immersion objective was used. For a series of experiments, the sample expected to give the brightest fluorescence was imaged first and the image was adjusted to the full dynamic range of the system. For all following samples of this series, these instrument settings were strictly maintained to allow the comparison of the intensities in all images within a series. Cells were observed in twenty successive focal planes, and images were processed with Leica Confocal Software (Leica Microsystems). The images showing fluorescence signal are maximum intensity projections of successive focal planes. Differential interference contrast images were made for comparison.
Results
Lysosomes Disappeared During Apoptosis of GC B-cells
GC B-cell apoptosis involves both caspase and cathepsin activity (Van Eijk and De Groot 1999; Van Eijk et al. 2003). Because cathepsins are generally localized in lysosomes, we decided to further explore the contribution of possible lysosomal leakage to apoptosis of GC B-lymphocytes.
Viable, freshly isolated GC B-cells stained with the lysosomal probe LTG displayed speckled staining patterns (Figure 1A). Based on the analysis of optical serial sections of individual cells obtained by CSLM, it was estimated that five to ten lysosomes were present in individual GC B-cells (data not shown). At 16-hr culture at 37C, GC B-lymphocytes were apoptotic, as shown by Annexin-V staining (Figure 1A). Concomitantly, the number of cultured GC B-cells that showed LTG staining was strongly reduced. Absence of LTG staining was observed in cells that stained for Annexin-V, suggesting a correlation between lysosomal destabilization and apoptosis.
Lysosomal leakage was an early event during GC B-cell apoptosis, because cells with decreased LTG staining were already found at 4 hr of culture (Figure 1B). At 16 hr of culture, the majority of the cells had lost lysosomal staining. Similar lysosomal destabilization was found after AO staining (CSLM data in Figure 1C; flow cytometry data in Figure 2A).
To further explore a possible role of lysosomal destabilization in GC B-cell apoptosis, we estimated the kinetics of lysosomal destabilization in relation to the kinetics of other apoptotic parameters. At 8 hr of culture, GC B-cells showed considerable lysosomal destabilization as well as several apoptotic features, including PS exposure, mitochondrial instability, caspase-3 activation, and DNA cleavage (Figure 2A). The kinetics of these parameters are shown in Figure 2B. The first signs of apoptosis were detected at 1 hr of culture. All parameters rapidly increased within the next 4 hr and subsequently leveled off. Lysosomal destabilization, as demonstrated by loss of AO red fluorescence, showed kinetics similar to the apoptotic parameters. Hence, it cannot be determined from these experiments whether lysosomal destabilization preceded the apoptotic process or was a consequence of apoptosis.
Lysosomal Destabilization Coincided With Cytoplasmic Cathepsin B Activity
To study the consequences of lysosomal destabilization, the activity of a major lysosomal proteinase, cathepsin B, was localized in living, freshly isolated B-cells and after 16 hr of culture (Figure 3). Cathepsin B activity was present in granular structures resembling lysosomes in freshly isolated B-cells. After 16 hr of culture, granular structures were hardly recognizable and most activity was found homogeneously throughout the cells, suggesting that activity was present in both cytoplasm and nucleus.
CD40 Ligation Prevented Lysosomal Destabilization
We and others have previously shown that the death receptor pathway is the main route of apoptosis in GC B-cells, and that CD40 ligation leads to silencing of the death receptor route by maintenance of cFLIPL in the DISC (Hennino et al. 2001; Lagneaux et al. 2001; Van Eijk et al. 2001a). Therefore, CD40 ligation provides a tool for determining whether lysosomal destabilization is under the control of the death receptor pathway or is an independent process.
Freshly isolated GC B-cells were cultured in the presence of an α-CD40 antibody and analyzed for lysosomal destabilization, cathepsin B activity, and apoptosis. In line with previous experiments, CD40 ligation suppressed apoptosis in GC B-cells in a dose-dependent fashion (Figure 4A). Moreover, CD40 ligation decreased lysosomal destabilization and leakage of cathepsin B from lysosomes in these cells (Figures 3 and 4A). The effects of CD40 ligation on the localization of cathepsin B activity varied rather strongly. Cells with intact granular structures containing cathepsin B activity were present as well as cells with completely ruptured lysosomes, as shown in Figure 3. GC B-cells cultured in the presence of an irrelevant isotype-matched control antibody (1 μg/ml) did not show any inhibition of apoptosis or lysosomal leakage (data not shown). These data indicated that lysosomal destabilization during apoptosis of GC B-cells was inhibited by CD40 ligation.
Lysosomal Destabilization Was Under Control of Caspase-8 Activity but Not of Caspase-9 Activity
To further unravel the relationship between the death receptor pathway and lysosomal destabilization, three different caspase inhibitors were tested for their ability to prevent PS exposure, lysosomal leakage, and cytoplasmic cathepsin B activity. These included the pan-caspase inhibitor ZVAD, the caspase-8-selective inhibitor IETD, and the caspase-9-selective inhibitor LEHD. ZVAD and IETD efficiently inhibited Annexin-V staining, lysosomal destabilization, and the presence of cathepsin B activity outside lysosomes, whereas LEHD had no significant effect (Figures 3 and 4B). These data indicate that lysosomal destabilization was not an autonomous process, but rather a result of caspase-8 activity in the death receptor pathway. These experiments did not support a significant role for caspase-9 activity in the induction of lysosomal destabilization in GC B-cells.
The Lysosomotropic Drug MSDH Bypassed the Death Receptor Route and Induced Apoptotic Features
The observation that lysosomal destabilization, extra-lysosomal cathepsin B activity, and apoptosis were blocked by inhibition of caspase-8 activity enabled us to isolate the effects of lysosomal destabilization from other caspase-8-dependent processes. By the addition of the lysosomotropic drug MSDH to GC B-cells in which caspase-8 activity was inhibited by IETD, the downstream effects of lysosomal disruption could be studied. As shown in Figure 5A, inhibition of caspase-8 activity by IETD resulted in maintenance of lysosomal LTG staining, even at 16 hr of culture. When IETD was combined with MSDH, the speckled lysosomal staining was clearly reduced, in agreement with the concept that lysosomes become disrupted by the addition of MSDH. The same phenomenon was observed with flow cytometry when lysosomes were stained with AO (Figure 5B). Remarkably, flow cytometry demonstrated that the addition of MSDH induced PS exposure and mitochondrial inactivation in IETD-treated cells without activation of caspase-3. MSDH treatment also induced significant formation of DNA strand breaks in these cells, but this effect was only partial within the time frame tested.
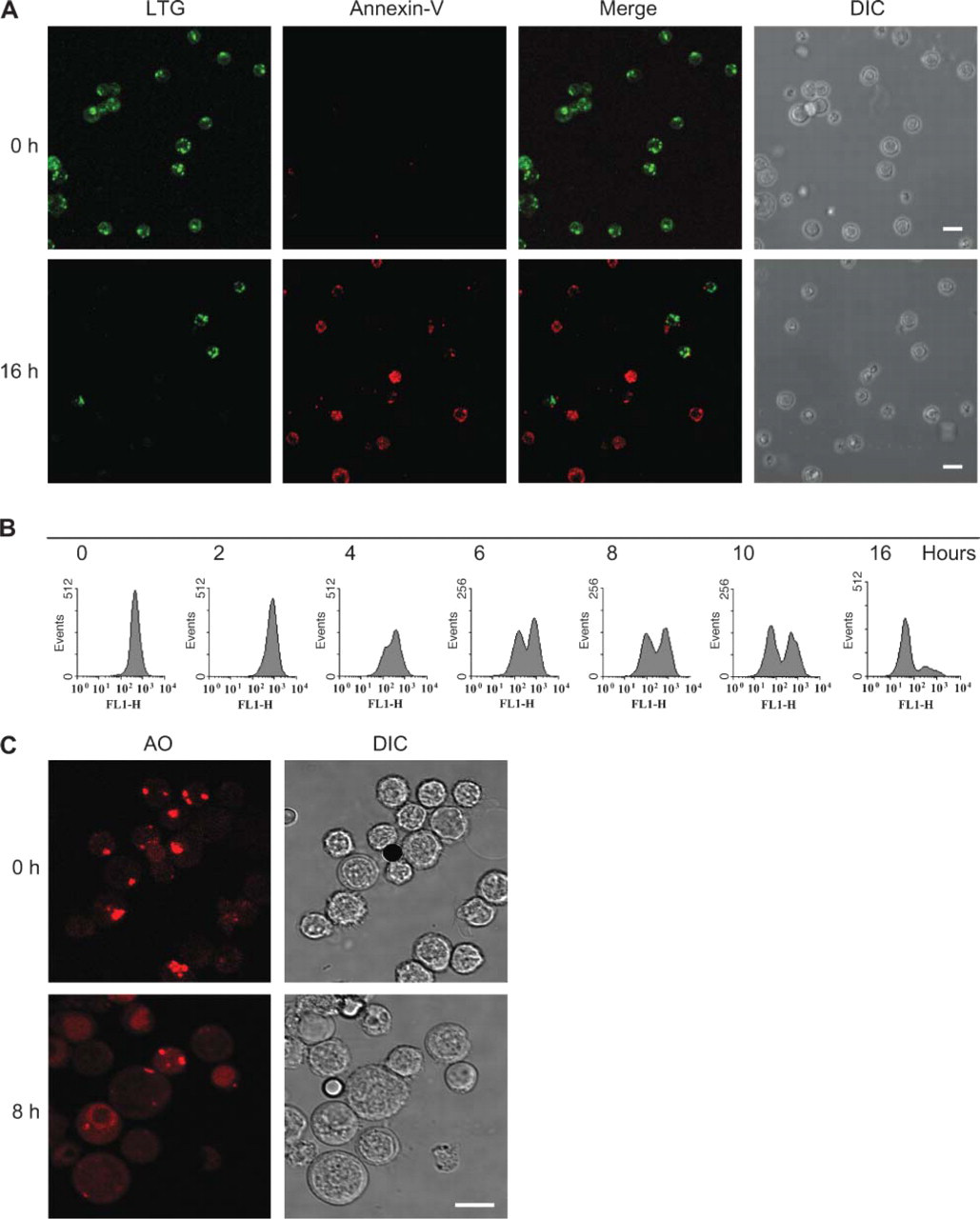
Lysosomal destabilization during apoptosis of germinal center (GC) B-cells. (
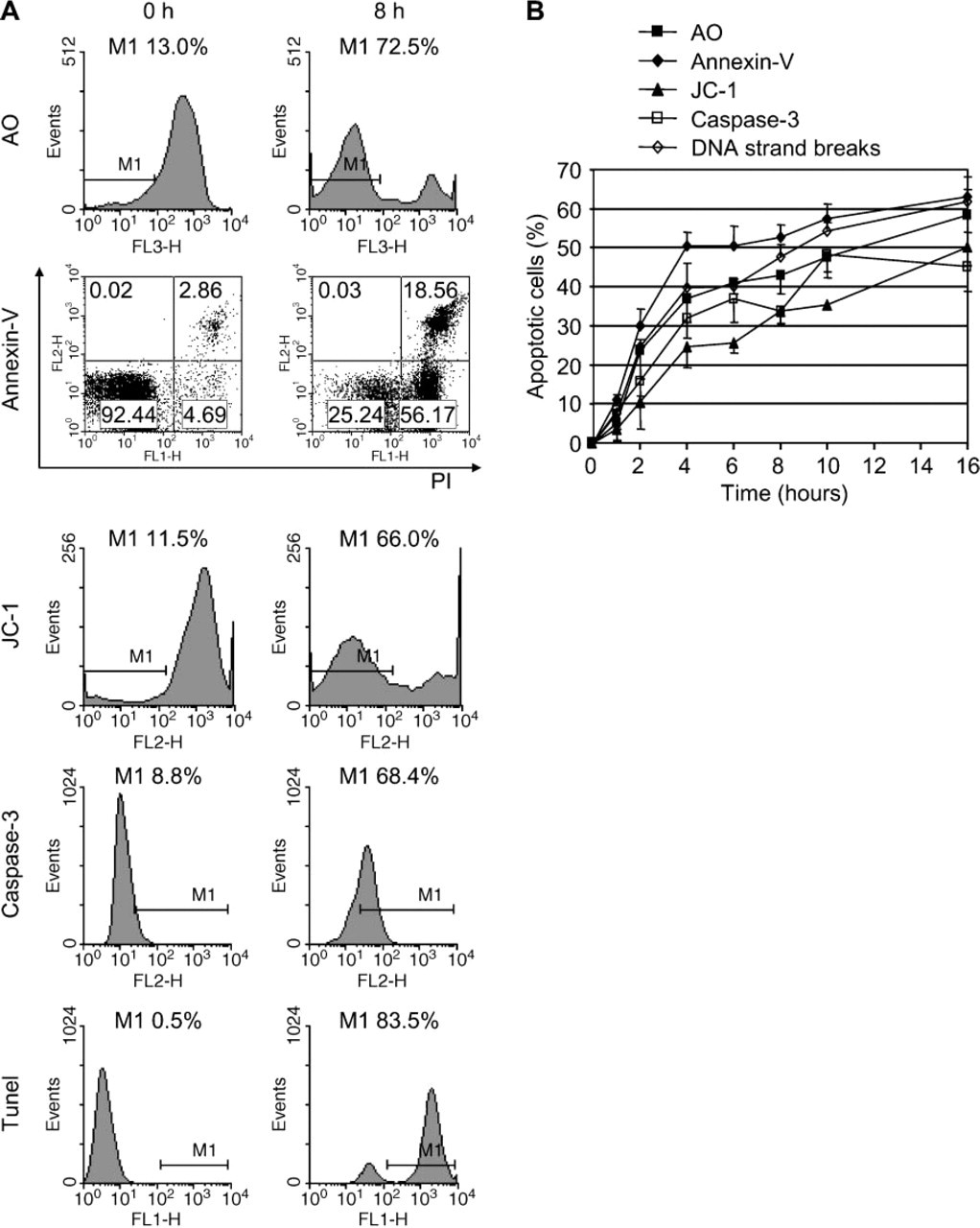
Kinetics of lysosomal destabilization and other apoptotic features in GC B-cells. (
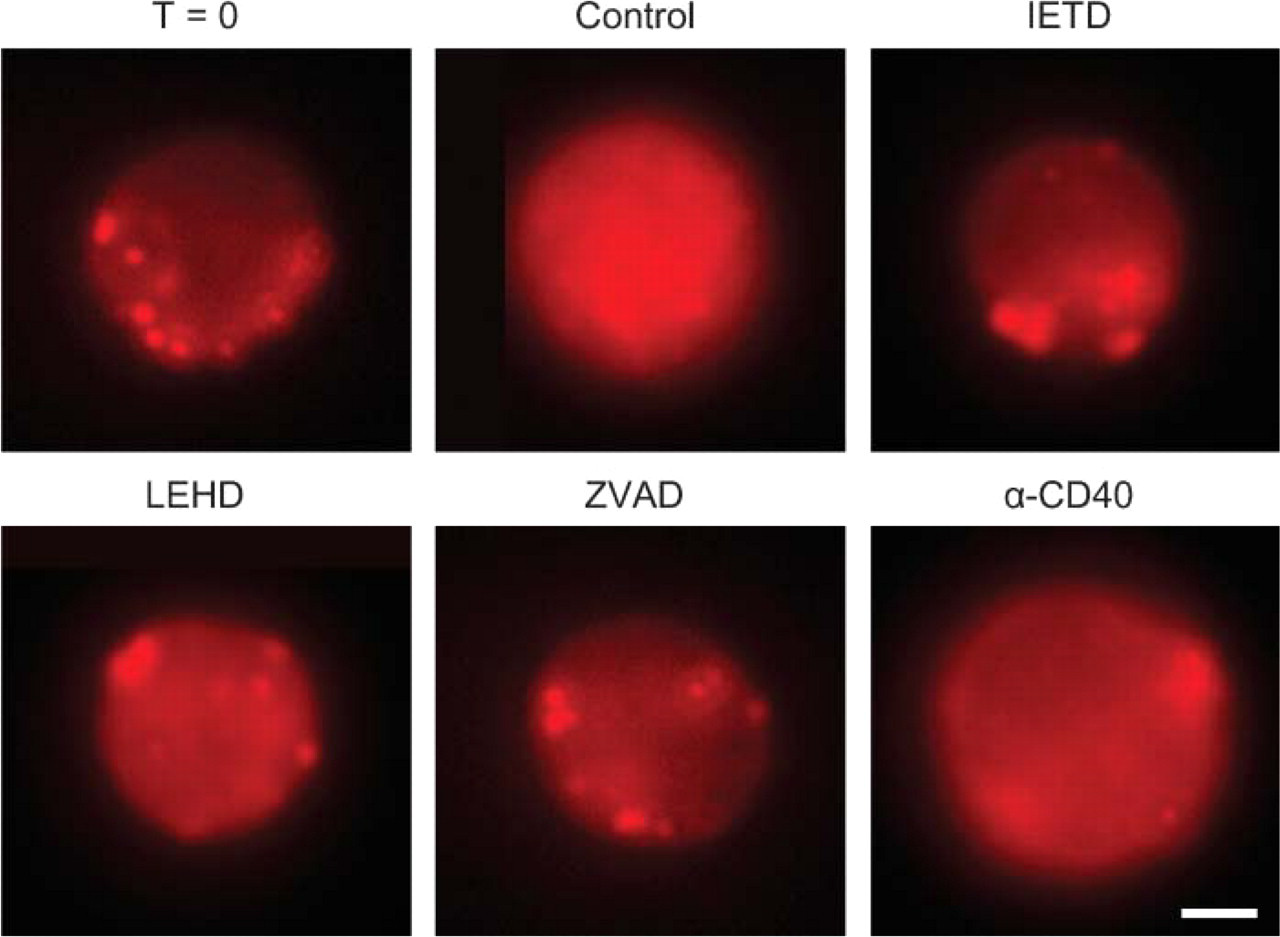
Active cathepsin B leaked from destabilized lysosomes in GC B-cells. Fluorescence images of live B-cells after incubation in a medium containing 2 μM selective fluorogenic cathepsin B substrate, [Z-Arg-Arg]2-cresyl violet. Incubation was performed directly after isolation of B-cells (T = 0) or after 16 hr of culture in the absence of any inhibition (Control) or in the presence of caspase-8 inhibitor (IETD), caspase-9 inhibitor (LEHD), pan-caspase inhibitor (ZVAD), or α-CD40 monoclonal antibody (MAb) (α-CD40). Granular (lysosomal) localization of cathepsin B activity was present in freshly isolated B-cells (T = 0), IETD-treated and ZVAD-treated cells, and, to a lesser extent, in α-CD40-treated cells, whereas a more homogenous extralysosomal distribution of cathepsin B activity was found in B-cells incubated in the absence of inhibitor (Control) and in LEHD-treated B-cells. Bar = 2 μm.
FDCs Protected GC B-cells Against Lysosomal Destabilization
Because we have shown previously that FDCs silence the death receptor route in adhering GC B-lymphocytes (Lindhout et al. 1993; Koopman et al. 1997; Van Eijk et al. 2001b), we have tested the possibility of inducing lysosomal disruption in FDC/B-cell clusters. Such clusters are spontaneously and rapidly generated upon in vitro culture of FDC-enriched GC B-cell fractions. FDC/B-cell clusters cultured for 16 hr at 37C showed intense lysosomal staining (Figure 6A) independent of the presence of the lysosomotropic drug MSDH (Figure 6B). Similarly, B-cells that were detached from FDCs after 16 hr of co-culture showed significantly increased lysosomal stability in comparison with cultured single B-lymphocytes (Figure 6B), implying that FDCs protected lysosomal stability in adhering B-cells. This protective effect of FDCs could be observed at up to 40 hr of culture (data not shown). Moreover, the lysosomotropic drug MSDH was unable to induce any lysosomal destabilization in B-cells in clusters of B-cells and FDCs in vitro (Figure 6B). This implies that FDCs protect adhering GC B-cells from apoptosis, not only by preventing caspase activation (Van Eijk et al. 2001a) but also by an efficient protection against lysosomal leakage.
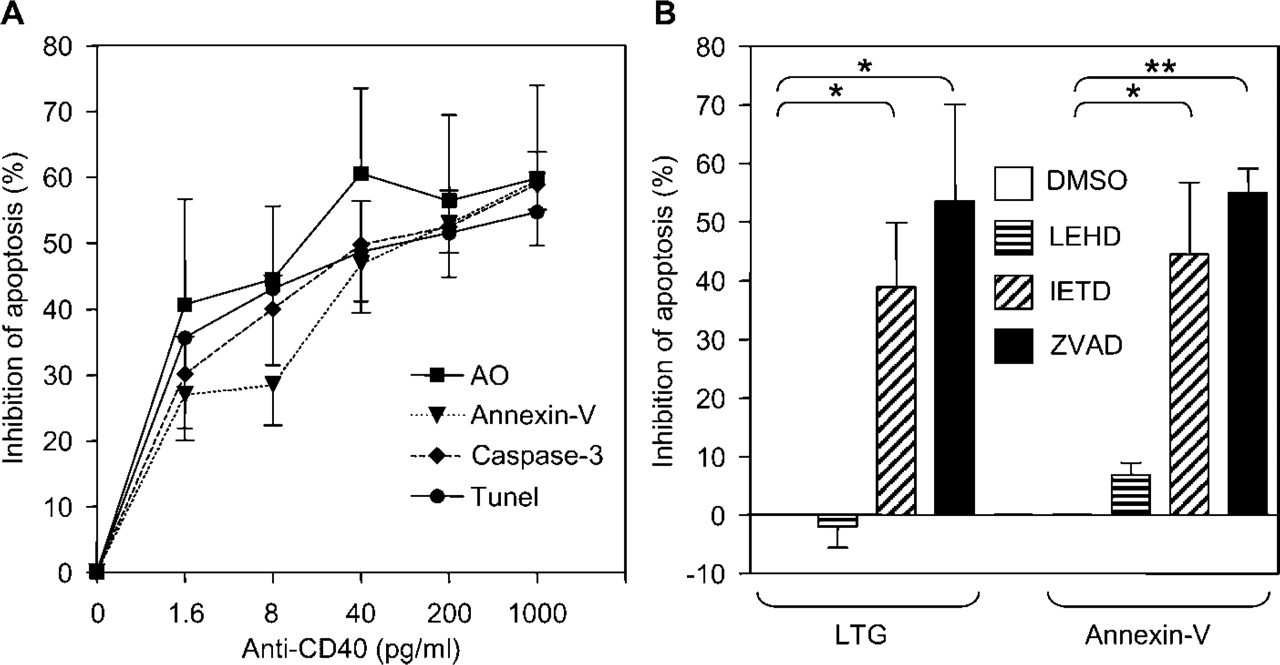
Lysosomal destabilization was under the control of caspase-8. (
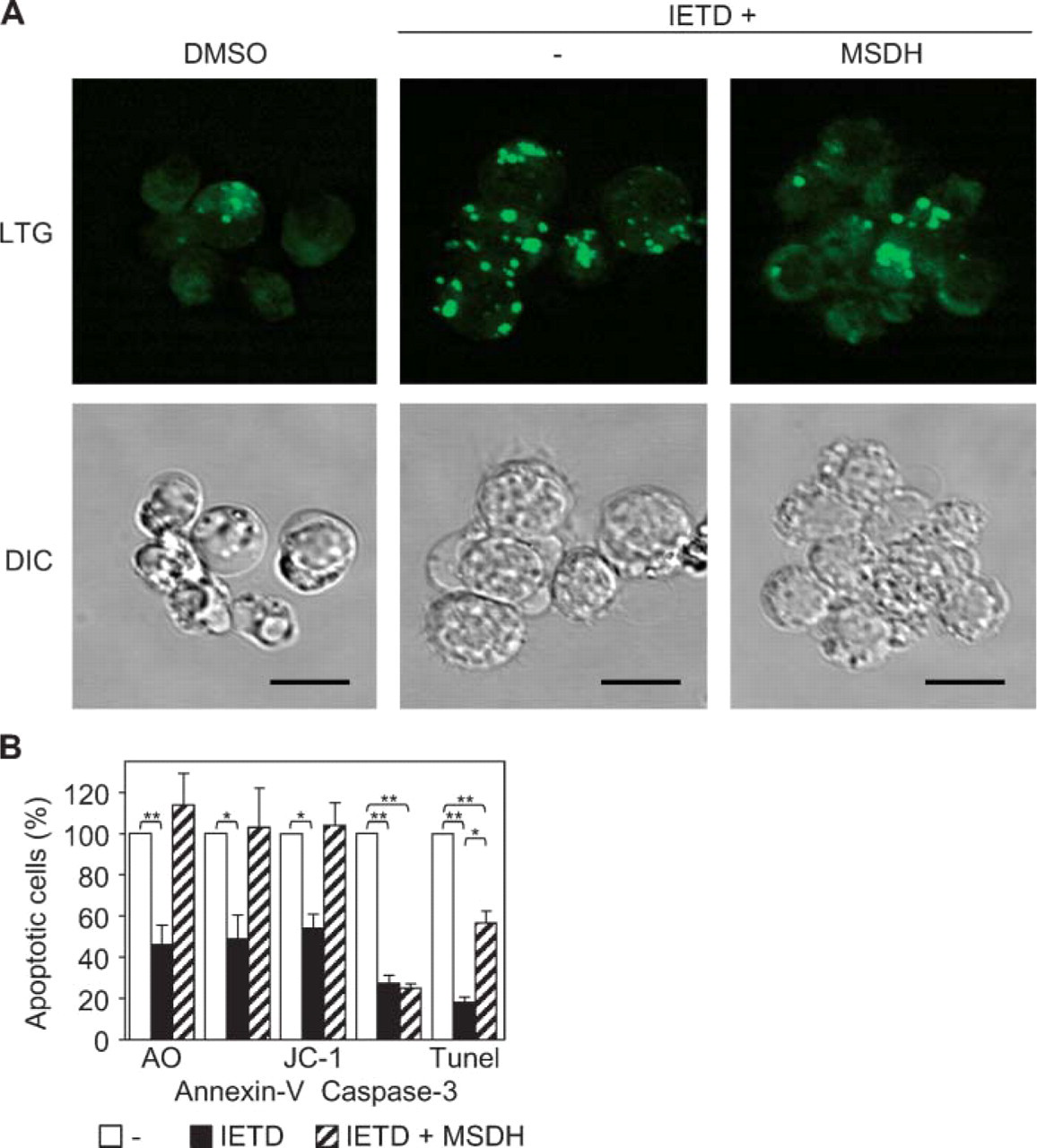
The lysomotropic drug O-methyl-L-serine dodecylamide hydrochloride (MSDH) induced apoptotic features without caspase activation. (
Discussion
The data presented here show that lysosomal destabilization and occurrence of activity of lysosomal hydrolases such as cathepsin B in cytoplasm and nucleus are early events during apoptosis of GC B-lymphocytes in vitro. Lysosomal leakage is under the control of caspase-8 activity and leads to several apoptotic features, including mitochondrial inactivation, PS exposure, and DNA cleavage.
Lysosomal destabilization has been shown to be an important phenomenon during apoptosis (Cossarizza et al. 1993; Jaattela and Tschopp 2003). Several authors have shown that lysosomal rupture occurs at or upstream of the level of mitochondrial inactivation (Guicciardi et al. 2000; Ferri and Kroemer 2001; Kagedal et al. 2001; Boya et al. 2003a,b), although exceptions have been noted as well (Michallet et al. 2004). A possible mechanism by which lysosomal rupture exerts its effect is by cleavage of Bid as a final result of mitochondrial leakage (Stoka et al. 2001; Reiners et al. 2002; Cirman et al. 2004).
In GC B-lymphocytes, the mitochondrial route of apoptosis seems less important, because inhibition of caspase-9 activity has a very limited effect on apoptosis (Hennino et al. 2001). Therefore, it is unlikely that lysosomal destabilization in GC B-cells is under the influence of mitochondrial inactivation. Indeed, by using caspase-8-specific and caspase-9-specific inhibitors, we have confirmed data (Hennino et al. 2001) that the caspase-8-dependent death receptor route is the dominant pathway that triggers apoptosis in these cells and that lysosomal leakage depends on this pathway. We have also shown that a consequence of lysosomal leakage is activity of lysosomal hydrolases such as cathepsin B in cytoplasm and nucleus. Cathepsin B has an optimum activity at acid pH, but it has a second activity peak at slightly alkaline pH (Mort and Recklies 1986; Van Noorden et al. 1998). This indicates that lysosomal destabilization in B-cells leads to hydrolytic activity in cytoplasm and nucleus. This caspase-8-dependent release of hydrolases from lysosomes into cytoplasm and nucleus may well be responsible for the cathepsin activity that is involved in the last proteolytic step in activation of DNA fragmentation (Van Eijk and De Groot 1999). It also explains why cathepsin-3 does not have to be activated for DNA fragmentation when lysosomes are disrupted, as we found in our experiments. It has been claimed by Van Eijk and De Groot (1999) that cathepsin-dependent DNA fragmentation is downstream of caspase-3. However, this conclusion was drawn on the basis of inhibition experiments using ZVAD and E64d, a pan-caspase inhibitor and a cathepsin inhibitor, respectively, with broad specificity. Therefore, it may well be that caspase-8-dependent lysosomal leakage is responsible for caspase-3-independent late activation events of apoptosis in GC B-cells, as was suggested by Van Eijk et al. (2003). This fast induction of apoptosis in GC B-cells that lack high-affinity BCRs to bind to FDCs and thus are not protected against apoptosis may have an important role in the prevention of autoimmune diseases, because B-cells with low or unwanted affinity BCRs may induce auto-immune reactions.
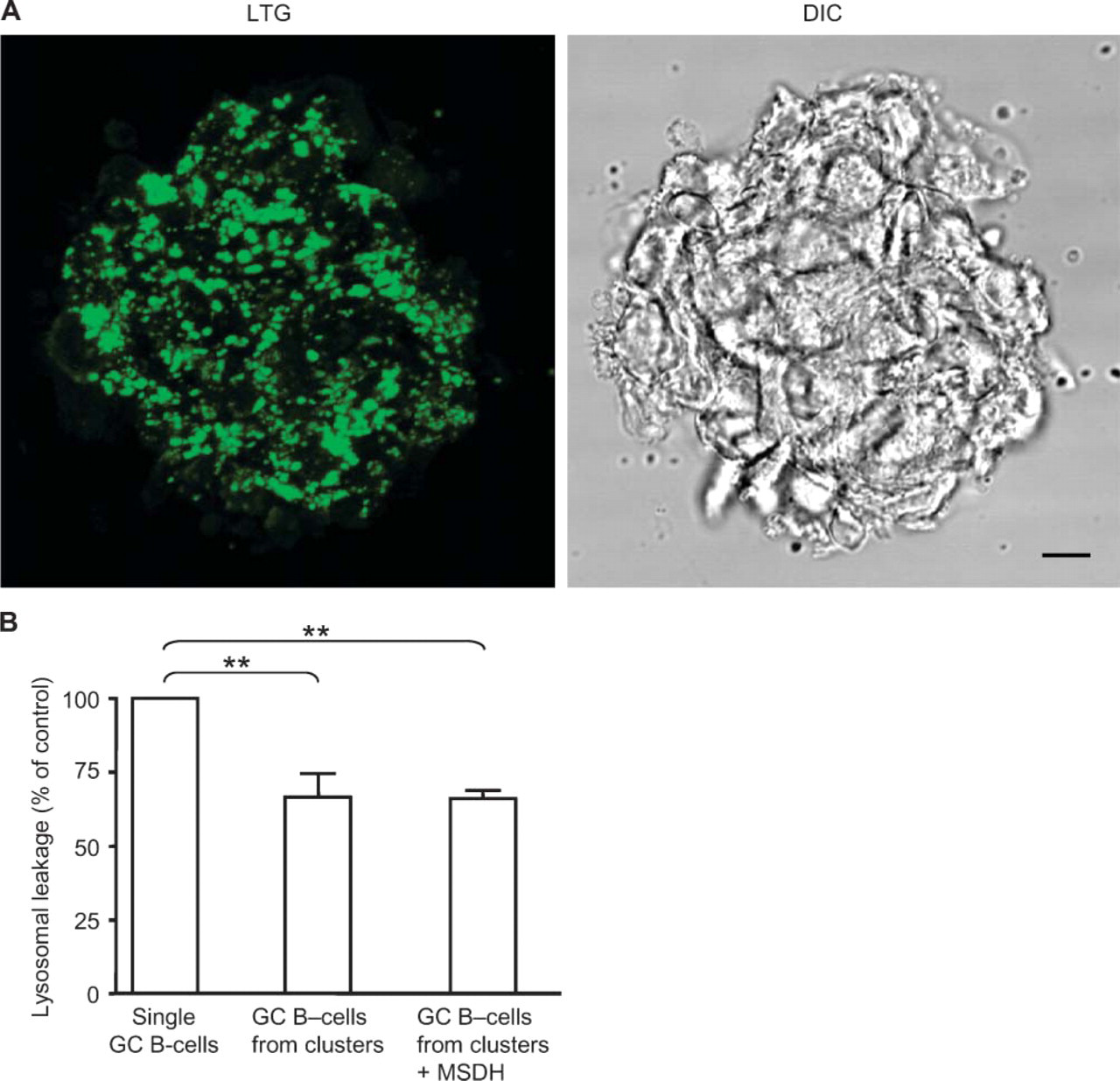
Follicular dendritic cells (FDCs) maintained lysosomal stability in adhering B-lymphocytes. (
CD40 ligation decreased lysosomal leakage to an extent similar to that of its inhibition of other elements of apoptosis. The evidence presented here indicates that lysosomal leakage is under the strict control of caspase-8 activity and does not occur as an autonomous process. Moreover, it shows that mitochondrial inactivation is not a significant trigger for lysosomal leakage in GC B-lymphocytes.
Because silencing of caspase-8 activity leads to stabilization of lysosomes, it was possible to study the isolated effects of lysosomal rupture by the addition of the lysosomotropic drug MSDH to GC B-cells that were treated with the caspase-8-selective inhibitor IETD. MSDH accumulates in lysosomes and induces leakage and translocation of cathepsins from lysosomes to the cytosol (Li et al. 2000; Boya et al. 2003b). In GC B-cells pretreated with IETD, addition of MSDH resulted in apoptosis, as demonstrated by increased Annexin-V staining. In addition, MSDH clearly induced mitochondrial inactivation as well as DNA fragmentation. Of note, all apoptotic parameters induced by this artificial lysosomal rupture occurred without any sign of caspase-3 activation.
In previous studies, we have shown that FDCs effectively protect adhering GC B-lymphocytes against apoptosis (Lindhout et al. 1993,1997) by preventing the dissociation of cFLIPL from the DISC and thus silencing the death receptor pathway in these B-cells (Van Eijk and De Groot 1999; Van Eijk et al. 2001a). In addition, FDCs switch off the endonuclease activity in GC B-cell nuclei (Lindhout et al. 1993,1995). Here we show that the protection by FDCs includes prevention of lysosomal destabilization (Figure 6).
In single GC B-lymphocytes treated with the caspase-8 inhibitor IETD, the lysosomotropic drug MSDH can bypass the blockade of caspase-8 activity and induce apoptotic features without involvement of caspase-3. However, MSDH has no effect on both lysosomal stability and other apoptotic features in B-cells that have established contact with FDCs. This indicates that the protective effect of FDCs includes more than just inhibition of caspase-8 alone. Therefore, we hypothesize that FDCs use mechanisms in addition to caspase-8 inhibition to prevent lysosomal instability in adhering GC B-lymphocytes. The molecular basis of this mechanism is presently unknown, but cystatin A, which is expressed at high levels in FDCs, may contribute here, as was proposed previously by Van Eijk et al. (2003).
Inhibition of caspase-8 can also result in cell death with autophagic features not only during development but also in cells of mature organisms (Yu et al. 2004). These authors demonstrated that caspase-8 inhibition induced autophagic features and subsequent death in fibroblasts. Autophagic cell death does not occur in human GC B-cells when caspase-8 is inhibited; otherwise, selection of B-cells with high-affinity BCRs by prevention of apoptosis due to FDC-B-cell interactions would not be possible. Moreover, autophagic cell death is a relatively slow process compared with apoptosis induced by lysosomal leakage, and thus would not fit in the process of rapid elimination of B-cells with low-affinity BCRs to prevent unwanted immune responses.
Footnotes
Acknowledgements
Part of this work was subsidized by Stichting De Drie Lichten. We thank the Onze Lieve Vrouwe Gasthuis hospital in Amsterdam for providing us with tonsils, Dr. Gene Dubowchik (Bristol-Myers Squibb; Wallingford, CT) for the kind gift of MSDH, Dr. Mark de Boer (Pangenetics; Utrecht, The Netherlands) for the α-CD40 antibody, and Dr. Jan van Marle for expert technical help with CSLM.