
Abstract
Autoimmunity is a well-established causative factor of premature ovarian failure (POF), and evidence for the same has been well reported in the literature. Detection of specific autoantibodies remains the most practical clinical research marker of any autoimmune disease. Variation in efficiency and specificity in the detection of ovarian autoantibodies has been reported. However, the frequency of false positivity and a solution to overcome this has not yet been reported. Herein, we report autoantibody to albumin as the likely responsible agent for false positivity. Our data indicate that presence of naturally existing autoalbumin antibodies in the circulation of normal women is responsible for the false signal seen in SDS-PAGE Western blot analysis and in immunohistochemistry (IHC). Having shown the presence of anti-albumin antibody in normal women as well as in the sera of POF patients, we have developed a novel blocking agent to overcome this problem. A high titer polyclonal antibody against human serum albumin was generated. This antibody showed immunoreactivity to albumin obtained from various sources. Preincubation of Western blots and IHC sections with this antibody drastically reduced background signals. The advantage of using this blocking was evident by identification of specific anti-ovarian antibodies in a group of POF patients. This blocking procedure made it possible to obtain a clear indication of the ovarian antibody status in women presenting with autoimmune POF.
Keywords
T
Diagnosis of AI-POF is confirmed by detection of ovarian autoantibodies in serum and is the most practical approach for the diagnosis of autoimmune diseases (Luborsky 2002). Vallotton and Forbes (1966) were the first to describe the presence of antibodies to rabbit ova cytoplasm using sera from POF patients.
However, there is great variation in the identification and incidence of ovarian autoantibodies reported among investigators. There could be several reasons for differences among study results. First, study design elements, such as antibody test format and antigen preparation and criteria for study and comparison groups, differ. Second, there may be several antigenic targets, and often only one could be assessed. Moncayo et al. (1990) developed an ELISA using microsomes from bovine corpora lutea as the antigen. Luborsky et al. (1990) developed an ELISA kit using total human ovary/oocytes as antigen and showed that sera from 71% of women with POF in their study had antibodies either to whole ovary or to oocytes. Wheatcroft et al. (1994) reported an incidence ranging from 24% to 60%, depending on the source of ovarian antigen. This difference was attributed to a cyclical variation in antigenic proteins. Using human ovarian tissue homogenate, Fenichel et al. (1997) carried out ELISA and showed a 59% incidence of anti-ovarian antibodies in patients with POF, of which the IgG isotype prevailed and was followed by IgM and finally IgA isotypes. However, at the present time there is no validated serum marker that can establish with certainty a diagnosis of AI-POF (Wheatcroft et al. 1997). Another major drawback in detection of ovarian antibodies in serum is the rate of false positives. A commercially available kit that tested for detection of anti-ovarian antibodies was reported to have poor specificity and tested positive in nearly one third of normally menstruating women (Novosad et al. 2003). One of the probable causes of nonspecificity could be due to the presence of naturally occurring antibodies (NAA). The presence of NAA has long been known. These NAA react to various cellular self-constituents. The major role of these NAA is in the regulation of the immune system. Their function in the first line of defense mechanisms has also been well elucidated in immunology textbooks. They are also known to play a role in the clearance of aging cells (Avrameas 1991). One of these NAA is the antibody that is directed against the ubiquitous protein albumin. Sansonno et al. (1986) suggested that anti-albumin antibodies (AAA) may have a possible role in the removal of effete albumin moieties. Louzir et al. (1992) demonstrated the presence of AAA in sera from 56 patients with hepatitis B-virus-related chronic liver disease and 30 normal individuals. Their results indicated that, regardless of their origin, autoantibodies are present in high amounts in the sera of individuals. Becketal. (1983) have also shown the presence of AAA directed against bovine serum albumin (BSA) in their study involving children with neuro-blastoma. These studies indicate the existence of NAA.
In the context of POF, a false positive report indicating autoimmunity as the mechanism of spontaneous POF could put young women at risk of inappropriate therapy, with serious consequences such as development of osteonecrosis due to glucocorticoid therapy (Kalantaridou et al. 1999). As the clinicians' diagnosis will be based exclusively and extensively on the detection of the presence or absence of ovarian antibodies, it is crucial that the diagnosis is foolproof. There is, therefore, a need to investigate the causes of nonspecificity and to improve the accuracy and efficiency of detection. A simple and specific test for ovarian antibodies would provide a useful serological marker in POF, enabling patients with ovarian autoimmunity to be identified early in the course of the disease. Consequently, they may have a chance of successful ovulations and thereby successful pregnancies.
In the present report we have determined the reason for false positives and have developed a simple but novel blocking method to overcome the nonspecific reactivity both in IHC and Western blot analysis with reproducibility. This simple, non-invasive, sensitive, and specific diagnostic test will aid the clinicians in correctly identifying patients with autoimmunity early in the course of the disease.
Materials and Methods
Experimental Subjects
The present study was performed with prior approval from the Institute's Clinical Ethics Committee. For this study, 25 women diagnosed with POF from April 2004 to March 2005 were recruited from the Reproductive Endocrinology and Infertility Clinic of the Institute. These patients presented with secondary amenorrhea with serum follicle-stimulating hormone (FSH) levels > 40 IU/liter. Serum FSH levels were checked on two different occasions with an interval of 1 month. All patients were <40 years of age at the onset of ovarian failure and had not had any ovarian and/or any other pelvic surgery (except for a few who had undergone diagnostic laparoscopy). Patients having genetic involvement, abnormal karyotypes, radiation/chemotherapy, viral oophoritis, or galactosemia-induced POF were excluded from the study. All subjects had an average height of 150 cm, well-developed secondary sexual characteristics, and no dysmorphic clinical symptoms.
Ten ml of blood was collected by routine venipuncture by well-trained laboratory technicians, and serum was separated. All serum samples were centrifuged and stored at −20C for further studies. Sera were coded and assessed without knowledge of corresponding medical information. Twenty healthy, normally menstruating women with a median age of 28 years served as controls. They had no evidence of any autoimmune disease or any history of infertility. Neither patients nor controls were receiving steroids at the time of blood sampling and collection.
Animals
Inbred male Belgium white rabbits (1-2 years old) and female Holtzman rats (15-20 weeks of age) were housed in a temperature-controlled room with a 12-hr light cycle. Animals were provided food and water ad libitum. All animal care practices and experimental procedures complied with the guidelines of the Care and Prevention Society against Cruelty of Experimental Animals (CPSCEA) on animal care and were approved by the Institutional Animal Ethics Committee.
Isolation of Total Protein from Ovarian Tissue
Procurement of human ovarian tissue of good quality and adequate quantity is difficult. Moreover, the entire experimental system involves human primary and anti-human secondary antibodies and, therefore, using an antigenic source from human origin would result in high background. Therefore, rat antigen was the source of choice. Ovaries used for isolation of total protein were collected from six sexually mature, untreated female rats. Animals were sacrificed by decapitation, and the ventral abdominal wall was cut open. Ovaries were separated from the oviduct-uterus complex, excess fat was trimmed off, and ovaries were washed twice in chilled PBS to remove traces of blood. Ovaries were then homogenized in distilled water containing 1% SDS (SISCO-SRL; Mumbai, India), 0.2% (3-[3-cholamidopropyl)dimethylammonio]-1-propane sulfonate (CHAPS; SISCO-SRL), and cocktail of protease inhibitors (Roche; Mannheim, Germany) using a polytron homogenizer (Kinematica AG; Lucerne, Switzerland). The homogenate was left overnight at 4C and sonicated (Ralsonex; Mumbai, India) the following day for 5 min on ice. The resultant homogenate was then cold centrifuged at 10,000 X g (Eppendorf 541 5R; Eppendorf AG, Hamburg, Germany) for 30 min and the supernatant collected, aliquoted, and stored at −20C. Total protein content of the ovaries was estimated by the modified method of Lowry using BSA as the protein standard (Lowry et al. 1951).
Production of Polyclonal Antibody to Human Albumin
Prior to immunization, sufficient preimmune blood was collected through the marginal ear vein of the rabbit. Serum was separated and stored at −20C until further use. Immunization protocol was carried out as described by Joshi et al. (2003) with slight modifications. Briefly, 100 μg of human serum albumin (HSA; Sigma, St Louis, MO) dissolved in 1 ml 0.01 M phosphate-buffered saline (PBS), pH 7.4, and mixed with an equal volume of Freund's complete adjuvant (Sigma) was injected subcutaneously around 30 sites. Two boosters at an interval of 15 days were given using Freund's incomplete adjuvant (Sigma), and postimmune serum was collected 10 days after the last booster, aliquoted, and stored at −20C. This postimmune serum containing the AAA was used for subsequent experiments. The AAA was titrated against HSA, rat serum albumin (RSA; Sigma), and BSA (Sigma) using ELISA and Western analysis.
Enzyme-linked Immunosorbent Assay
The protocol was carried out as described by Khole et al. (2000). Briefly, 96-well microtiter plates (Nunc; Roskilde, Denmark) were coated with HSA, RSA, and BSA in separate modules at a concentration of 1 μg/100 μl using carbonate-bicarbonate coating buffer, pH 9.6, by incubating at 4C overnight. Excess solution was flicked off the next day and modules were washed with PBS containing 0.05% Tween-20 (Sigma). Nonspecific binding sites were blocked with 5 g% non-fat dry milk (Nakoda; Mumbai, India) in PBS (NFDM-PBS) for 1 hr at room temperature. Then, 100 μl of the postimmune serum (polyclonal to albumin) at serial dilutions from 1:1000 to 1:400,000 in 2 g% NFDM-PBS was added to the wells and incubated at 37C for 1 hr in duplicate. Preimmune serum was used at 1:100. Unbound antibodies were removed by washing wells six times with PBS containing 0.05% Tween-20 (Sigma). One hundred μl of suitably diluted (1:3000) horseradish peroxidase (HRP)-labeled swine anti-rabbit secondary antibody (Dako; Glostrup, Denmark) in 1 g% NFDM-PBS was added and incubated at 37C for 1 hr. The wells were then washed as described above and immunoreactivity was visualized using 200 μl of 3,3′5,5′ tetra-methylbenzidine/hydrogen peroxide (TMB/H2O2) substrate (20X; Bangalore Genei, Bangalore, India) prepared by adding 500 μl TMB/H2O2 + 9.5 ml distilled water in dark. Reaction was stopped by adding 100 μl of 4 N H2SO4 (Qualigen; Mumbai, India) and optical density measured at 450 nm on a Titerteck multiscan plate reader (Bio-Tek Instruments; Winooski, VT).
SDS-PAGE and Western Blot Analysis
Forty μg of total rat ovarian (RO) protein or 5 μg each of HSA, RSA, and BSA were loaded per well in the loading buffer containing β-mercaptoethanol and heat denatured at 95C for 5 min on a 10% SDS-PAGE. Proteins were separated by electrophoresing at 80 V for 3 hr as described by Laemmli (1970). Separated proteins were then electrophoretically transferred to nitrocellulose membranes (Amersham Biosciences; Piscataway, NJ) under cooling conditions according to the procedure described by Towbin et al. (1979) using an electroblotting apparatus (BioRad Laboratories; Richmond, CA) at 80 V for 2 hr. Transferred proteins were visualized using Ponceau S (Sigma). Individual lanes were appropriately marked, isolated, and blocked for 2 hr at room temperature with NFDM-PBS or NFDM-PBS containing 20% AAA. Serum from controls or patients was added to the strip and incubated at 4C overnight. A strip with no primary antibody served as ‘secondary only’ control. Membrane strips were washed the following day three times with 0.1% Tween 20 in PBS and then incubated with suitably diluted (1:100,000) HRP-labeled goat anti-human secondary antibody (Sigma) in 1 g% NFDM in PBS for 1 hr at room temperature. Strips were then washed as described above. Detection was done using enhanced chemiluminescence plus kit (ECL; Amersham Biosciences) as per standard protocol with luminol serving as substrate.
Modified ‘Lunch Box’ Immunoadsorbent Technique
This protocol was carried out as described by Rybicki (1986) with slight modifications in accordance with our experimental conditions. It consists of a subtractive technique, whereby a protein of interest (preferably at high concentration) is blotted onto a membrane electrophoretically and then used for immunoadsorptive purification of a specific antibody from serum. This kind of procedure could readily be used to purify specific antibodies for a protein. A total of 300μg of HSA (per 20-mg well) was electrophoretically separated in a reducing environment and transferred onto a nitrocellulose membrane for 1 hr. The blot was stained with Ponceau S, and the region covering the 66-kDa protein was cut from the rest of the blot and then destained in D/W. The blots were directly incubated with a fixed volume of the serum from controls as well as from patients and agitated at 4C for a period of 36, 60, and 130 hr. After each point of incubation, the ‘immunoad-sorbed’ serum was collected, labeled carefully, and stored at −20C until further use. All time point ‘immunoadsorbed’ sera were then evaluated for reactivity using SDS-PAGE Western blot analysis using RO as the antigen. In brief, 40 μg of ovarian protein was electrophoretically transferred onto a nitrocellulose membrane, stained to visualize bands with Ponceau S and then blocked with NFDM-PBS for 1 hr at room temperature. Immunoadsorbed serum from controls as well as from patients was then added to the membrane strips and incubated at 4C overnight in a moist chamber. The following day, the strips were washed three times with 0.1% Tween 20 in PBS and then incubated with suitably diluted (1:100,000) HRP-labeled goat anti-human secondary antibody (Sigma) in 1 g% NFDM in PBS for 1 hr at room temperature. Strips were then washed as described above and detected using ECL kit.
Immunohistochemistry
IHC was carried out as per the protocol described by Khole et al. (2000). In brief, rat ovaries were excised, cleared of fat and blood, washed with chilled PBS, and fixed for 48 hr at room temperature in Bouin's fixative prepared by mixing 105 ml of saturated picric acid (Qualigens), 35 ml formaldehyde (Qualigens), and 7 ml glacial acetic acid (SISCO-SRL). Tissues were then dehydrated through serial grades of alcohol, cleared in xylene (Qualigens,) and embedded in paraffin (Qualigens). Five-μm-thick serial sections were cut on a microtome (RM 2125RT; Leica, Wetzlar, Germany). Only those sections containing follicles at different stages were taken so that most stages in folliculogenesis could be viewed at a glance. Paraffin from sections was melted at 56C and deparaffinized in xylene. Endogenous peroxidase activity was quenched with methanol containing 0.03% H2O2 and rehydrated through a graded series of alcohol solutions in decreasing concentrations. Sections were then blocked with either 5 g% BSA in PBS, 20% normal goat serum (NGS-PBS; Vector Laboratories, Burlingame, CA) in PBS, NFDM-PBS, or NFDM-PBS containing 10% AAA or 20% AAA separately, for 2 hr at room temperature. Serum from control or patients was added to the slide and incubated at 4C overnight in a humid chamber. The section serving as ‘secondary only’ control was blocked in NFDM-PBS and then incubated with 100 μl of 2.5 g% NFDM in PBS instead of primary antibody for the same period. The following day, slides were washed three times with PBS for 10 min each. They were then incubated with suitably diluted (1:1000) goat anti-human secondary antibody (Sigma) in 1 g% NFDM in PBS for 2 hr at room temperature in a humid chamber. After washing three times with PBS for 10 min each, the immunoperoxidase color reaction was developed using 3,3′-diaminobenzidine (DAB) substrate chromogen solution (Sigma), which was prepared by adding 0.003% H2O2 to 0.08% solution of DAB in PBS (Qualigens) and color was developed in the dark for not more than 10 min. The reaction was stopped by immersing the slides in D/W for 5 min and then counterstained for 30 sec using Delafield's hematoxylin (Qualigens). The sections on the slide were allowed to differentiate under running tap water for 15 min, dehydrated through a series of alcohol grades, cleared in xylene, and mounted in DPX mounting medium (SRL; Mumbai, India). Slides were examined on a Zeiss axioscope microscope (Carl Zeiss Inc.; San Marcos, CA) at X 400 magnification.
Results
The main goal of this work was to devise a simple protocol for blocking nonspecific reactivity.
Sera from Women Show Antibody Targeted to a 66-kDa Protein
SDS-PAGE Western blot analysis using control as well as POF patients' sera revealed reactivity at about 66 kDa in all serum samples analyzed (Figure 1, Lanes C1 to P1). Apart from the 66-kDa protein, sera from POF patients (Figure 1, lane P1) targeted a protein lying in the region of 45-55 kDa. The ‘secondary only’ control (Lane NC) did not show any reactivity at this locus, thus confirming the specificity of the reaction obtained at the 66-kDa locus. The 66-kDa locus is well known to constitute the ubiquitous protein albumin. Immunore-activity to this locus as seen above led us to hypothesize that there are autoantibodies to albumin in the general circulation.
Reactivity at the 66-kDa Locus Is Due to Anti-albumin Antibodies
To prove our hypothesis, we conducted an experiment, i.e., SDS-PAGE Western blot analysis where albumin from various sources as well as from RO was blotted, blocked with NFDM-PBS, and probed with sera from controls and patients. On the Western blot, control as well as patient sera revealed immunoreactivity to albumin from all sources as well as to a 66-kDa locus in the RO lane, which aligned to the loci of the albumins from other sources (Figure 2A). A ‘secondary only’ control did not show any reactivity to any of the antigens (Figure 2B). This experiment confirmed our hypothesis that women have AAA in their circulation and this AAA acts as an interfering agent in the discrete detection of ovarian-specific antibodies in AI-POF. Using ELISA, we observed that serum from normal women and normal men, as well as POF patients, showed an obvious difference in antibody titers when HSA was used as the coating antigen (data not shown).
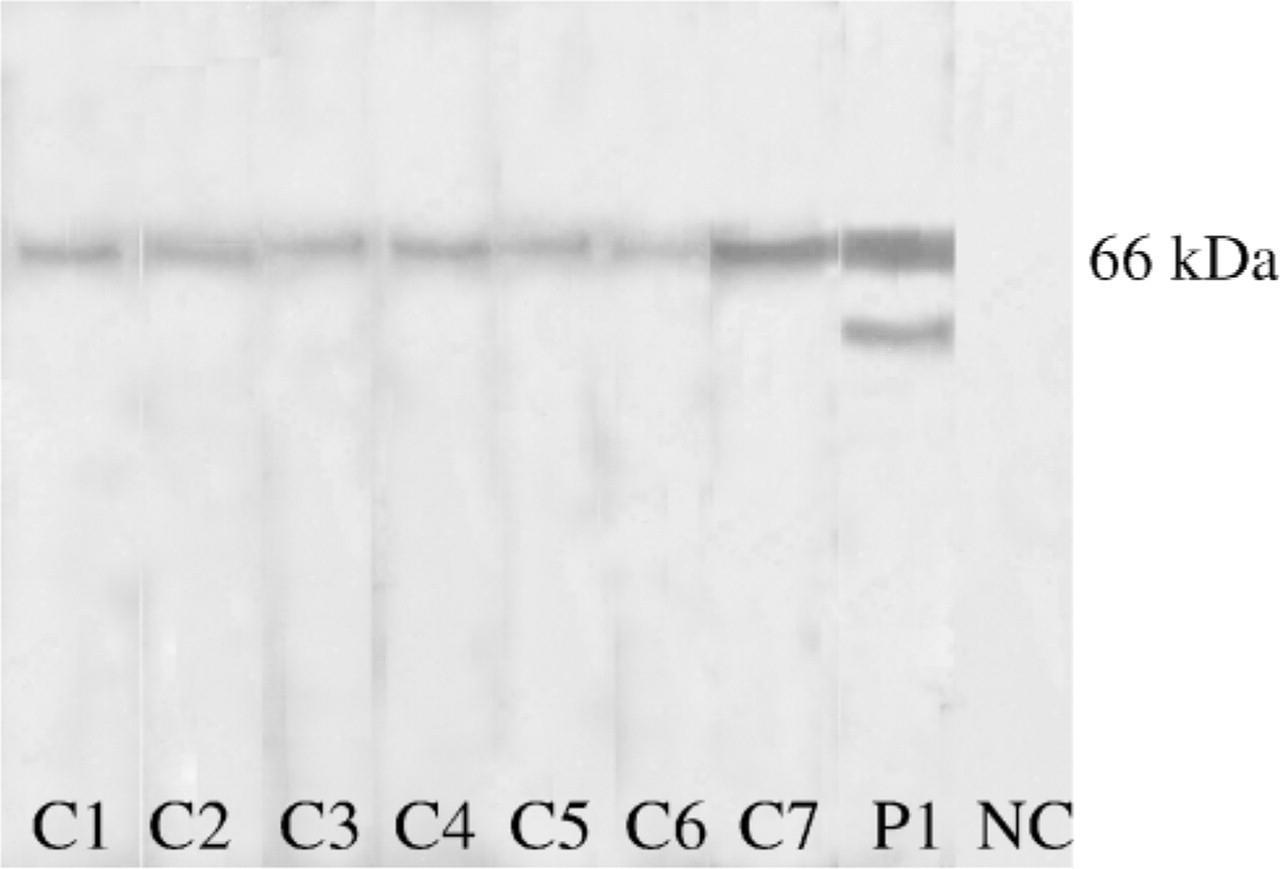
Western blot analysis of rat ovarian antigen using sera from control and POF patients. Notethatall sera from controls (C1 to C7) as well as patients (P1) react with a 66-kDa albumin locus. However, serum from patients also reacts to an antigen at the 45-to 55-kDa locus. A no primary antibody control (NC) shows no immu-noreactivity at the same locus.
Immunoadsorption of Serum with Albumin Is Not Effective in Specific Detection of Ovarian Antibodies
To remove the nonspecific reactivity, we used a ‘lunch box’ protocol, which is a simple, reliable “low-tech” procedure. We made certain modifications in this procedure for our experimental convenience as described in Materials and Methods. Eluates were collected at time points of 36, 60, and 130 hr, and at 36 hr there was no obvious reduction in the titer of AAA in control as well as in patient sera. Specific reactivity of patient sera also persisted. However, at 60 and 130 hr of incubation, reactivity toward the albumin loci was lost when both control and patient sera were used. But the specific reactivity to bands other than the 66 kDa seen earlier with patient serum was also lost (data not shown). The technique has been shown to be effective for isolating specific antibodies but was not useful for solving the problem. Moreover, as titer of AAA varies among individuals, feasibility of this technique seems unrealistic as standardization will have to be done for each serum sample. Therefore, we decided to block endogenous albumin rather than to make the serum AAA free and for this reason decided to raise polyclonal antibodies to albumin.
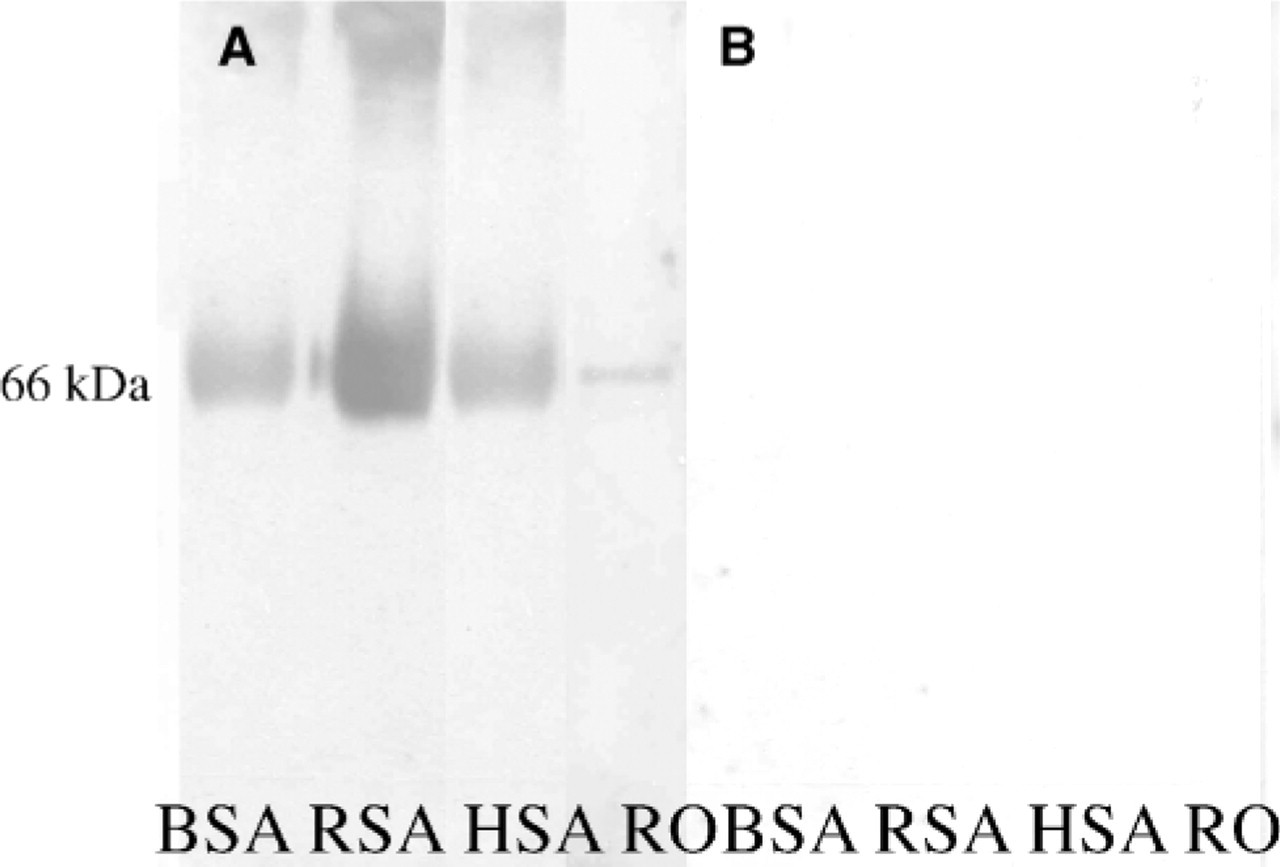
Sera from controls as well as from POF patients show the presence of anti-albumin antibodies reacting to the 66-kDa protein by Western blot analysis. (A) Nonspecific reactivity using control serum at 66-kDa albumin locus using 5 g% NFDM-PBS as blocking solution is shown. (B) No immunoreactivity to any of the antigenic sources is shown when a secondary alone control was used. BSA, bovine serum albumin; RSA, rat serum albumin; HSA, human serum albumin; RO, rat ovarian antigen.
Production and Characterization of the Albumin Polyclonal Antibody
A polyclonal antibody to albumin was raised in rabbit and was titrated against HSA, RSA, and BSA by ELISA. Titer of the polyclonal antibody obtained was 1:280,000 against HSA, 1:28,500 for RSA, and 1:24,000 for BSA (Figure 3).
The rabbit preimmune serum at a 1:5000 dilution does not react to albumin from any source (Figure 4A). At the same dilution, the polyclonal antibody strongly reacted to the 66-kDa loci of HSA, RSA, and BSA and also to RO (Figure 4B). This indicated that the antibody recognizes albumin not only from serum source but also from tissue origin (Figure 4B, Lane RO). This polyclonal antibody was further used as a blocking agent to block all endogenous albumin moieties both in Western blot as well as in IHC.
Novel Blocking Aids Identification of True Histological Targets by Blocking Endogenous Albumin and Appreciable Reduction of Nonspecificity
Using a novel blocking method with incorporation of a 20% rabbit polyclonal AAA in NFDM-PBS, reactivity with the control serum at the albumin locus was appreciably reduced as demonstrated with HSA, RSA, and BSA and the RO albumin locus (Figure 5A). Moreover, it is important to note that the specific reactivity seen at the 45- to 55-kDa region using patient serum was not lost. The nonspecific reactivity at the albumin locus seen using patient serum (Figure 5B) was completely removed on employing the novel blocking protocol (Figure 5C). Thereby, the novel blocking method serves as a better alternative than the immunoadsorption method to remove nonspecific reactivity with the naturally occurring human AAA.
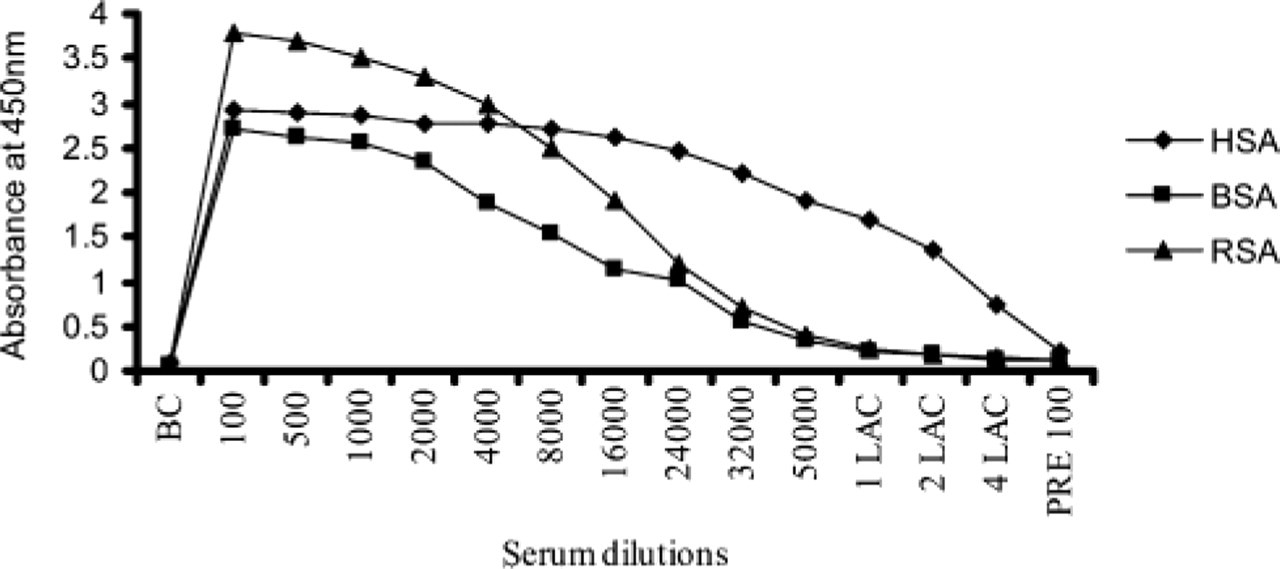
Titration of the polyclonal antibody raised to HSA using ELISA against the antigens. Titer of the polyclonal antibody obtained using HSA was 1:280,000; RSA was 1:28,500, and BSA was 1:24,000. Pre-100 is 1:100 dilution of the rabbit serum (preimmune serum) before immunization with HSA. 1:1 Lac corresponds to a dilution of 1:100,000 of dilution buffer, etc. BSA, bovine serum albumin; RSA, rat serum albumin; HSA, human serum albumin.
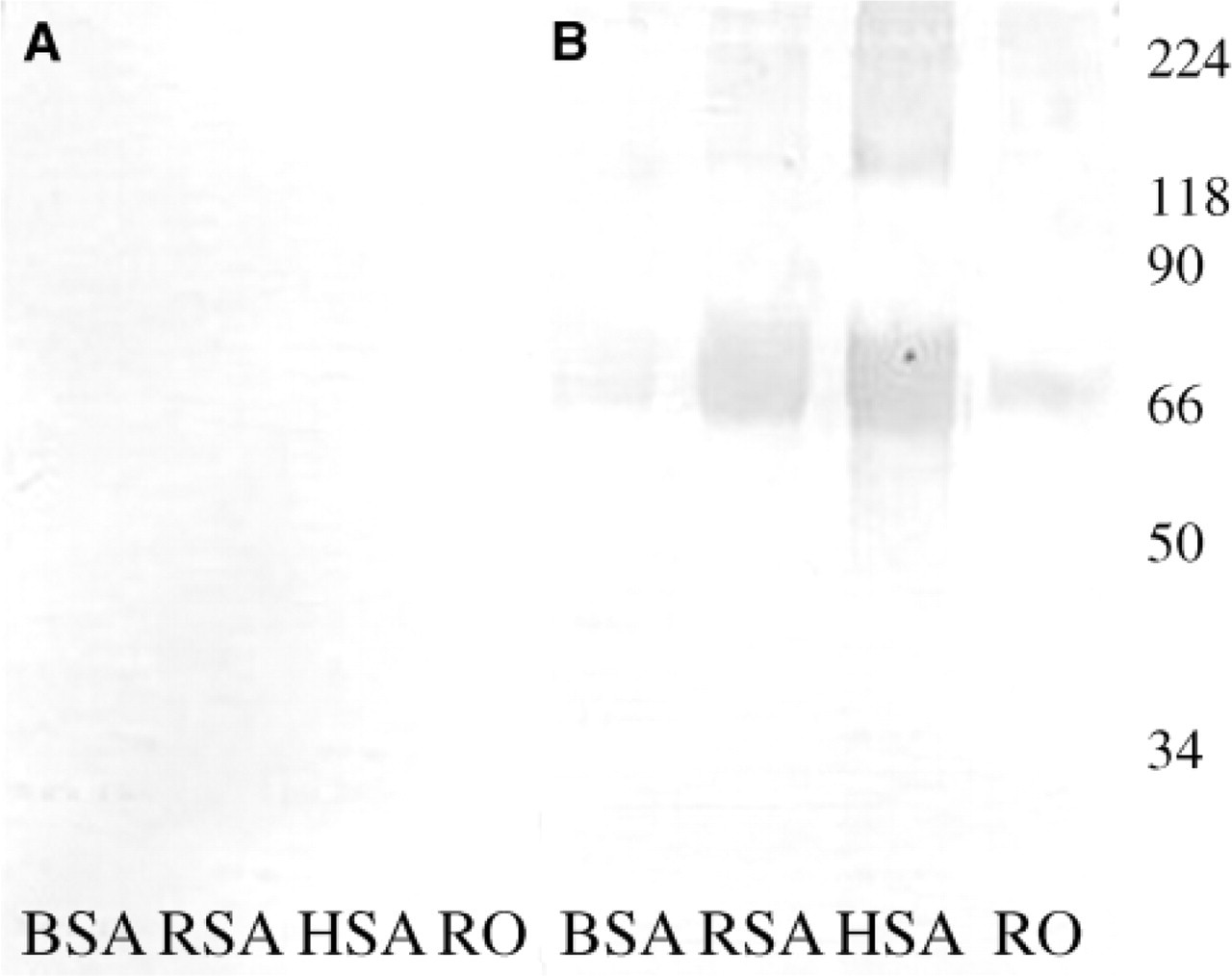
Western blot analysis of various antigens using rabbit polyclonal antibodies raised toHSA. (A) No immunoreactivity using a 1:5000 dilution of the preimmune serum is shown with any of the antigens used. (B) Reactivity to the 66-kDa albumin loci using the immune serum atthe same dilution as the preimmune serum against the same antigens. BSA, bovine serum albumin; RSA, rat serum albumin; HSA, human serum albumin; RO, rat ovarian antigen.
Using IHC, sera of normal women showed reactivity to the ovary section specifically to the oocyte when NFDM-PBS (Figure 6A) was employed for blocking. Different blocking protocols were employed to investigate the authenticity of the positive staining observed using serum from controls. Blocking with 5 g% BSA-PBS and 20% NGS-PBS showed no decrease in the intensity using sera from controls (Figures 6B and 6C, respectively). The ‘secondary only’ control did not show any reactivity with any of the ovarian components (Figure 6D) thereby ruling out staining due to nonspecific binding of secondary antibody. Interestingly, blocking with 10% (Figure 6E) or 20% AAA in NFDM-PBS (Figure 6F) resulted in disappearance of the nonspecific staining, with 20% AAA blocking being most effective. Thus, 20% AAA in NFDM-PBS was employed as blocking solution for further experiments using patient's sera. Using NFDM-PBS as blocking solution in IHC, there was nonspecific staining in all regions of the ovary with intense staining of the oocyte when patient serum was used. Sera from POF patients not only showed stronger reactivity to oocytes but also reacted to other ovarian cell types such as thecal cells and luteal cells, sparing the zona pellucida and granulosa cells (Figure 6G). However, after blocking with 20% AAA in NFDM-PBS, positive staining in the oocyte was still retained (Figure 6H). We therefore concluded that it was a specific staining. Those POF patients who were antibody negative by Western blot analysis showed no immunostaining when 20% AAA in NFDM-PBS was used for blocking, clearly showing that all nonspecific sites were appreciably blocked (data not shown).
Discussion
A review of the literature reveals that currently there is no clinically proven sensitive and specific serum test to confirm autoimmune involvement in POF. Reports from various investigators support the fact that there is antibody-mediated attack to the ovary in women with POF, but due to the lack of specific tests the marker antigen/s responsible for the autoimmune attack remains elusive.
Novosad et al. (2003) reported poor specificity with a commercially available kit, which tested positive in nearly one third of normally menstruating women. Our studies also indicate the existence of nonspecific reactivity to ovarian antigen. We conducted experiments revealing that one of the most predominant autoantibodies in the serum of controls as well as in patients is toward albumin moieties. These antibodies occur naturally and are present in most animals and humans where they may or may not have any pathological significance. A similar finding was observed by Sansonno et al. (1986) who developed an ELISA to detect antibodies to albumin in the sera of patients with acute hepatitis B disease. The authors reported the presence of IgG and IgM isotype antibodies to albumin in the sera of the 28 normal subjects enrolled in the study, although patients with the disease had significantly lower levels of the autoantibody to albumin.
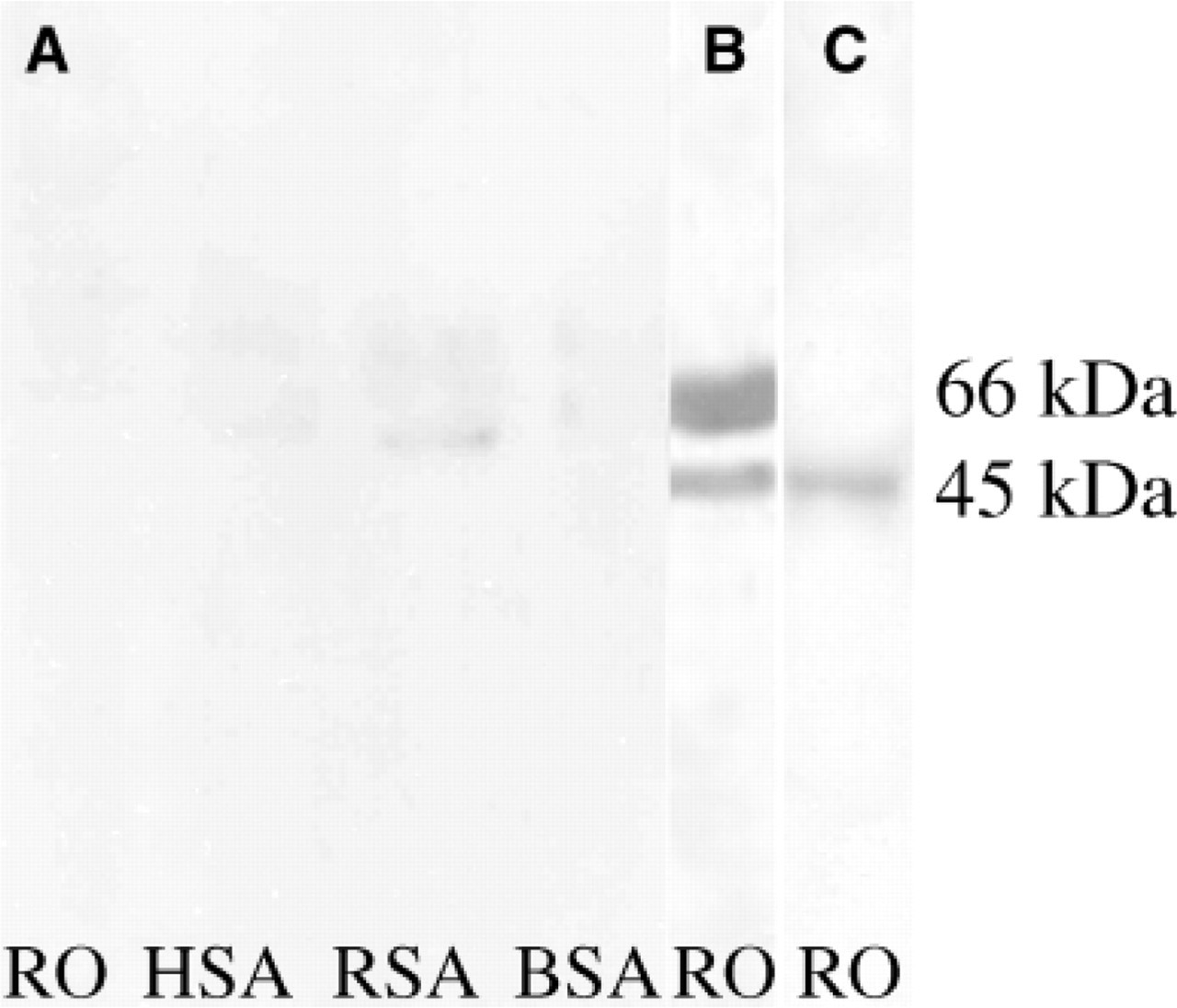
Western blot analysis of different antigens using sera from control and POF patients employing a novel blocking protocol. (A) Disappearance of the nonspecific reactivity using control serum when 20% AAA in 5 g% NFDM-PBS is used as blocking solution. (B) Nonspecific reactivity using patient serum at 66-kDa albumin locus using 5 g% NFDM-PBS as blocking solution. Apart from the nonspecificity at the albumin locus, a specific antigenic target at the 45- to 55-kDa locus was also observed. (C) Disappearance of the nonspecific reactivity when 20% AAA in 5 g% NFDM-PBS is used as blocking solution. However, using patient serum the specific reactivity at the 45- to 55-kDa locus is not lost, ensuring that the reactivity at this locus is not because of a cross-reactive protein but due to a specific antigenic target. BSA, bovine serum albumin; RSA, rat serum albumin; HSA, human serum albumin; RO, rat ovarian antigen.
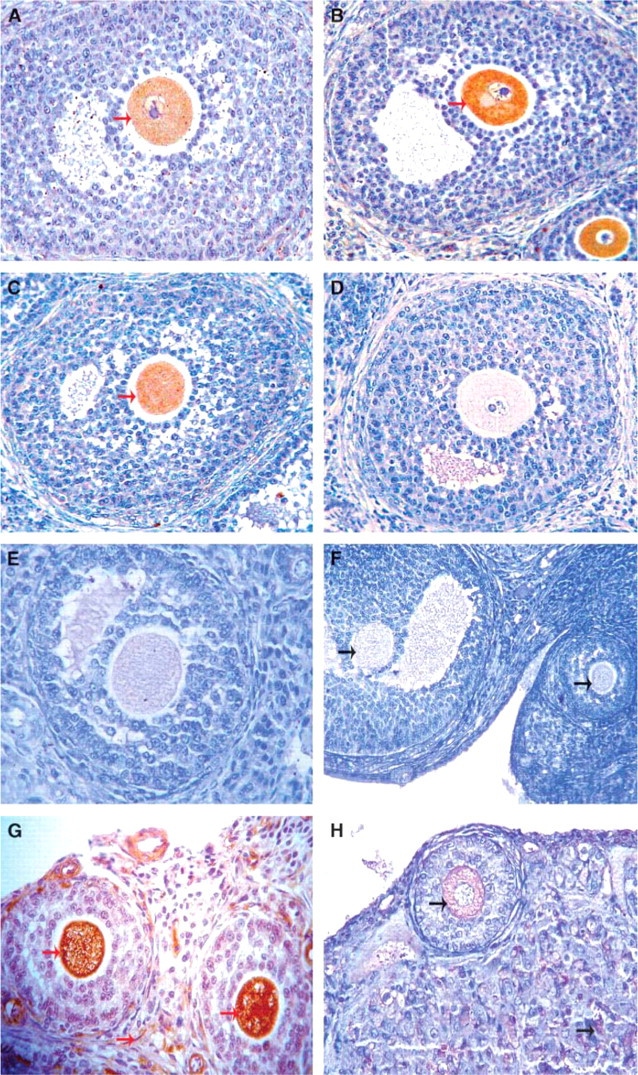
Immunohistochemistry using serum from controls as well as from patients against RO staged at the secondary follicle. No immunostaining is seen in (D) when secondary only control was used. Note that the persistence of immunostaining of the oocyte (nonspecificity) could not be blocked using different blocking solutions, namely, (A)5 g% non-fat dry milk (NFDM-PBS), (B) 5 g% BSA-PBS, (C) 20% normal goat serum (NGS-PBS). Effective blocking of this nonspecific reactivity could be achieved partially as seen in (E), 10% rabbit polyclonal anti-albumin antibody (AAA) in 5 g% NFDM-PBS and completely as seen in panel (F), 20% AAA in 5 g% NFDM-PBS. (G,H) Reactivity shown when using POF patient serum before and after using 20% AAA in 5 g% NFDM-PBS novel blocking solution, respectively. Red arrows indicate immunore activity to the oocyte before novel blocking and green arrows indicate immunoreactivity after employing novel blocking. Magnification: X 400.
The presence of antibodies to albumin could have a normal physiological function rather than manifestation of any disease, probably for the removal of effete albumin moieties in the serum (Sansonno et al. 1986). Albumin is conserved across species, irrespective of the source, namely, in the serum or from tissue. The mere presence of the antibody toward albumin will definitely give a false positive finding in tests used to detect anti-ovarian antibodies. Blocking the sites on albumin by using AAA raised in a different species prevented the binding of naturally occurring AAA in the serum, thus permitting the binding of ovarian-specific antibodies to their targets.
This hypothesis was tested by conducting SDS-PAGE Western blot analysis where we incubated the blot with sera from controls as well as from patients toward HSA, RSA, and BSA. Sera from both groups reacted to albumin from all three sources, thus providing strong and conclusive evidence of the existence of naturally occurring AAA in women. The next obvious step was to eliminate the reactivity of NAA to albumin. We had two choices available: a) to deplete albumin from tissues (this would aid mostly in soluble protein analysis), but for actual localization of the targeted antigen it is almost impossible to eliminate albumin in the ovarian sections by IHC; b) to achieve this, the second choice was to immunoadsorb the serum with albumin, which did not provide satisfactory results. Therefore, we decided to raise polyclonal antibodies to HSA and used this antibody as a novel blocking solution. Using the novel blocking solution in Western blot analysis where different sources of albumin and total RO were probed with control serum, the difference in reactivity toward albumin loci, in comparison to the blot where the blocking was not employed, was appreciable. There was loss of reactivity to albumin in the blot with the novel blocking solution. Using the same blocking regimen in IHC experiments with control and patient sera as probe, we were able to demonstrate the loss of nonspecific reactivity toward the oocyte employing 20% AAA in NFDM-PBS; however, specific reactivity using sera from patients was retained.
Monnier-Barbarino et al. (2005) discussed indirect immunofluorescence (IIF) and ELISA as two major approaches to detect ovarian antibodies in patients' sera and from other biological fluids. They have argued that the insufficient specificity and lack of reproducibility of IIF technique was due to the heterogeneity of the tissue sections. On the other hand, the same group preferred ELISA because it allows screening of a large panel of antigens and usually yields more homogeneous results than IIF.
However, based on our experimental observations, we feel that unless the titer of a particular antibody is extremely high and also due to poor representation of the cognate antigen, the test is not always decisive. For example, in the present study we have demonstrated that sera from most of the POF patients react to oocyte antigens. As this component of the ovary comprises a miniscule portion in comparison to other cell types, titers against these antigens in total ovarian protein would not be significant when ELISA is used to detect the antibodies. Additionally, in doing so, one is likely to miss the detection of antibodies to the oocyte antigens and report patient sera to be negative for ovarian antibodies. From our data, we also feel that IHC as a technique using tissue sections yields more information about the likely histological targets involved in POF, which would not be possible using either ELISA or SDS-PAGE Western blot analysis. It also enables the localization of antibodies within different compartments of the ovary and thus the determination of their histological targets. Moreover, with the incorporation of the blocking protocol that we introduced, a clear scenario of antibody status is presented and assists the clinician in accurate diagnosis and subsequent patient treatment. In the present study we evaluated a simple buteffective method for blocking nonspecific binding. Our group thus could provide a solution to the problem discussed by Novosad et al. (2003) regarding the reliability of the existing tests available to detect antibodies in women with AI-POF.
In summary, we have presented a simple method for blocking nonspecific binding and thus developed a specific test to detect AI-POF. Our results show that blocking of albumin with 20% AAA in NFDM-PBS will provide specific reproducible results not possible with the available diagnostic tests. Having established a specific non-invasive diagnostic test, we are one step closer to the identification of target antigens involved in ovarian autoimmunity. This will enable us to understand the immunological mechanisms responsible for POF. This observation could also have applications in other autoimmune diseases, where diagnosis is hampered due to lack of specific tests.
Footnotes
Acknowledgments
E.S.P. acknowledges the Lady Tata Memorial Trust, Mumbai, India for financial support (as a Junior Research Fellow) received during the course of the study. This study was financially supported by the Indian Council of Medical Research, New Delhi, India.
The authors thank Dr. Chander P. Puri, Director of the Institute, for support during the present study. We also thank Mr. Manish N. Ghosalkar for technical support.