
Abstract
Green fluorescent protein (GFP) expression was evaluated in tissues of different transgenic rodents—Sprague-Dawley (SD) rat strain [SD-Tg(GFP)Bal], W rat strain [Wistar-TgN(CAG-GFP)184ys], and M mouse strain [Tg(GFPU)5Nagy/J]—by direct fluorescence of native GFP expression and by immunohistochemistry. The constitutively expressing GFP transgenic strains showed tissue-specific differences in GFP expression, and GFP immunohistochemistry amplified the fluorescent signal. The fluorescence of stem/progenitor cells cultured as neurospheres from the ependymal region of the adult spinal cord from the GFP SD and W rat strains was assessed in vitro. After transplantation of the cells into wildtype spinal cord, the ability to track the grafted cells was evaluated in vivo. Cultured stem/progenitor cells from the SD strain required GFP immunostaining to be visualized. Likewise, after transplantation of SD cells into the spinal cord, immunohistochemical amplification of the GFP signal was required for detection. In contrast, GFP expression of stem/progenitor cells generated from the W strain was readily detected by direct fluorescence both in vitro and in vivo without the need for immunohistochemical amplification. The cultured stem/progenitor cells transplanted into the spinal cord survived for at least 49 days after transplantation, and continued to express GFP, demonstrating stable expression of the GFP transgene in vivo.
For transplantation studies, it is important to be able to unequivocally identify and track the transplanted cells in the host tissue. The most suitable approach to labeling cells for subsequent transplantation studies depends on a number of criteria, such as the retention of the label, which is important for long-term studies; whether the protocol to detect the label is compatible with other analyses, such as immunohistochemical determination of the phenotype of grafted cells; and whether the label is stably incorporated or has the potential to transfer to endogenous cells. The gene-based reporter green fluorescent protein (GFP) fulfills all these criteria and is therefore an ideal marker for labeling cells for transplantation.
GFP was originally derived from the bioluminescent jellyfish Aequorea victoria, and requires only light excitation for its autofluorescence (Tsien 1998). The visualization of GFP is non-invasive, and it can be quantified by flow cytometry, confocal microscopy, and fluorometric assays (Hadjantonakis and Nagy 2001). Cells can be pre-labeled in vitro by transfection with viral vectors containing the GFP gene, and detection of the fluorescent protein is dependent upon the stable and sustained expression of the transgene in transplanted cells. However, rates of transfection are often low and GFP expression can be significantly downregulated over time in transplanted cells (Onifer et al. 1993; Vroemen et al. 2003). A more sustained expression of the transgene in grafted cells in vivo is obtained when transplanted cells are derived from transgenic animals. Also, the problem of toxicity associated with viral vector delivery of the transgene is not a concern (Tucker 2001).
The objectives of this study were to evaluate in vivo GFP expression in neural and non-neural tissues of different GFP transgenic rodents; to culture stem/progenitor cells from the ependymal region of the adult spinal cord of the GFP rat strains and assess their fluorescence in vitro; and subsequently to transplant the cultured cells into wild-type rat spinal cord and evaluate the ability to track grafted cells in situ. Several mutants of the original wild-type GFP gene with improved thermostability and fluorescence have been engineered, including the enhanced GFP (EGFP) variant. The transgenic rodents utilized in this study all harbor the EGFP gene under the control of a constitutive promoter, and will be henceforth referred to simply as GFP. Two different GFP transgenic rat strains were used in this study, and GFP expression patterns were compared with a GFP transgenic mouse strain. GFP transgenic rats of Sprague-Dawley (SD) background were used [SD-Tg(GFP)Bal] that contained a lentivirus EGFP transgene driven by ubiquitin-C promoter and cytomegalovirus (CMV) enhancer (Lois et al. 2002). In the present study, this strain is referred to as the SD transgenic. We also examined hemizygous EGFP transgenic rats of Wistar background [Wistar-TgN(CAG-GFP)184ys]. This strain was originally generated by Kobayashi and colleagues (Hakamata et al. 2001), and carries the EGFP transgene driven by chicken β-actin promoter and CMV enhancer (Hakamata et al. 2001). This transgenic rat strain is referred to as the W strain in the present study to distinguish it from the SD rat strain described above. The hemizygous GFP transgenic mouse strain used [Tg(GFPU)5Nagy/J] also carries EGFP driven by chicken β-actin promoter and CMV intermediate early enhancer (Hadjantonakis et al. 1998). In the present study, this transgenic mouse line is designated the M strain. This is the first report to compare GFP transgenic rat strains for neural stem/progenitor cell transplantation studies.
Materials and Methods
Animals
The GFP transgenic SD rat strain [SD-Tg(GFP)Bal] was obtained from the NIH-funded Rat Resource and Research Center (RRRC), University of Missouri, Columbia, MO. The W rat strain [Wistar-TgN(CAG-GFP)184ys] is available from the YS Institute, Inc., Utsunomiya, Tochigi, Japan. The transgenic mouse M strain used [Tg(GFPU)5Nagy/J] was obtained from Jackson Laboratory, Bar Harbor, ME. For the transplantation experiments, wild-type SD rats (Charles River; St. Constant, Quebec, Canada) were used as transplant recipients of the cultured GFP cells.
All animal procedures were performed in accordance with the National Guide to the Care and Use of Experimental Animals (Canadian Council on Animal Care) and approved protocols from the Animal Care Committee of the Research Institute of the University Health Network, Toronto, Ontario, Canada.
Fluorescence Detection of Native GFP Expression in Tissues of Transgenic Rodents
The GFP transgenic rats or mice were sacrificed at the age of 6–8 weeks. Animals were deeply anaesthetized with an intraperitoneal injection of sodium pentobarbital and were transcardially perfused with 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS), pH 7.4. Tissue was cryoprotected in 30% sucrose in 0.1 M PBS for at least 1 week, then embedded in Shandon Cryomatrix compound (VWR International; Mississauga, Ontario, Canada) and cryosectioned into 20-μm sections on Superfrost slides (Fisher Scientific; Ottawa, Ontario, Canada). Slides were stored at - 80C until ready to be viewed and then thawed at room temperature; sections were rehydrated with PBS buffer. Fluorescent tissue was examined using a Nikon Eclipse TE 300 microscope (Nikon; Mississauga, Ontario, Canada), and images were captured with a CCD camera and Bioquant Software (R and M Biometrics, Inc.; Nashville, TN).
Culturing of Spinal Cord Stem/Progenitor Cells Derived from GFP Rats
Neurospheres were generated based on the methods of (Yamamoto et al. 2001), with several modifications. Adult male GFP rats (2 months old) were sacrificed, and the cervical spinal cord was excised and washed in 4C Dulbecco's phosphate-buffered saline (DPBS) supplemented with 30% glucose (Sigma-Aldrich; Oakville, Ontario, Canada). The overlying meninges and blood vessels were removed, and the white matter surrounding the central canal was removed. Only the tissue immediately surrounding the central canal, including the closely apposed ependymal and subependymal regions, was harvested, under sterile conditions. The dissected tissue was placed in fresh DPBS-glucose, cut into 1-mm3 pieces, and then transferred to the papain dissociation system enzyme solution (Worthington Biochemicals; Lakewood, NJ) consisting of 0.01% papain and 0.01% DNase I. The tissue was incubated in the enzyme solution at 37C for 1 hr with continuous agitation. Following incubation, the tissue was triturated with a 10-ml pipette and the enzymatic reaction stopped with ovomucoid protease inhibitor. The cell suspension was centrifuged for 5 min at 1000 rpm using a discontinuous density gradient to remove cell membrane fragments. Cells were resuspended in Neurobasal-A medium (Gibco-Invitrogen; Burlington, Ontario, Canada) supplemented with B27 (Gibco-Invitrogen), hormone mix, L-glutamine (Gibco-Invitrogen), penicillin/streptomycin (Gibco-Invitrogen), 20 ng/ml epidermal growth factor (Sigma-Aldrich), 20 ng/ml fibroblast growth factor-2 (Sigma-Aldrich), and 2 μg/ml heparin (Sigma-Aldrich). Viable cells were counted in a hemocy-tometer using the trypan blue exclusion assay. Cells were seeded at a density of 20 cells/μl in Nunc T25 culture flasks (VWR International), and cultures were maintained at 37C in an incubator with 100% humidity and 5% CO2. Neurospheres were passaged every 5–7 days. Passaging of neurospheres was accomplished by collecting the cell culture medium containing the free-floating neurospheres in a 50-ml centrifuge tube and centrifuging for 1 min at 1500 rpm. The supernatant was discarded, the pellet was resuspended in fresh supplemented Neurobasal-A medium, and the cell suspension was triturated 40 times using a 1-ml pipette. A live cell count was performed on a hemocytometer using the trypan blue exclusion assay, and cells were plated at a density of 20 cells/μl in Nunc T25 culture flasks and incubated at 37C in an incubator with 100% humidity and 5% CO2. Native GFP expression in cultured cells was examined under 488-nm excitation light (Nikon Eclipse TE 300 microscope).
Transplantation of GFP-expressing Spinal Cord Stem/Progenitor Cells into Wild-type Rat Spinal Cord
Wild-type adult female SD rats (250–300 g; Charles River) were used as transplant recipients of the cultured donor cells derived from the transgenic GFP rat strains. Rats were anesthetized by inhalation of 5% halothane, which was reduced to 2% during surgery, in combination with a mixture of nitrous oxide and oxygen (1:2, v/v). The spinal cord was exposed by laminectomy at thoracic level T8, and a small opening in the dura was made with a 30-gauge needle at the injection site to aid penetration of the micropipette tip into the spinal cord. Neurospheres in passage 3 (4–7 days in vitro) derived from the GFP rat spinal cord as described in the above section were dissociated and resuspended in culture medium at 10,000 cells/μl. Cell viability was assessed with trypan blue exclusion, and 5 μl of the cell suspension was injected at a rate of 2.5 μl/min via a 100-μm-aperture glass micropipette into the intact spinal cord at level T8. With the aid of the operating microscope, the micropipette was stereotactically inserted into the cord 1 mm from the midline using the dorsal midline vein as a landmark, and at a depth of 1 mm. Following injection, the micropipette was left in place for 2 min to prevent back-flow of cells. To aid transplant survival and integration, animals were immunosuppressed daily until sacrifice with 10 mg/kg of cyclosporine (Sandimmune, Novartis; Dorval, Quebec, Canada) injected intraperitoneally. Rats were sacrificed 1 day, 3 days, and 49 days after transplantation by lethal overdose with pentobarbital and transcardially perfused with 4% paraformaldehyde in PBS. Tissue was cryoprotected in 30% sucrose, and a rostrocaudal segment of the spinal cord 1.0 cm in length encompassing the transplant site was removed, embedded in OCT compound, and cryosectioned longitudinally into 20-μm serial sections.
GFP Immunostaining
To enhance native GFP expression in vitro and in vivo, cultured cells and transgenic tissue or spinal cord sections containing GFP-grafted cells were immunostained with a polyclonal anti-GFP antibody (Abcam, Inc.; Cambridge, MA). GFP immunocytochemistry on cultured cells was performed as described by Jasinska et al. (1999). Cells were incubated for 40 min at 4C with 5% CO2 in supplemented Neurobasal-A medium containing 0.1% bovine serum albumin (BSA) (w/v) with 1:2500 anti-GFP antibody (Abcam, Inc.). After washing three times with supplemented Neurobasal-A medium containing 0.1% BSA and twice with supplemented Neurobasal-A medium alone at 4C, cells were fixed with acetone/methanol (1:1) for 2 min at room temperature for permeabilization. Cells were then washed with PBS and incubated with anti-GFP antibody (1:2500) in 0.1% BSA for 40 min at 4C. After washing twice with supplemented Neurobasal-A medium and 0.1% BSA for 20 min, cells were incubated with goat anti-rabbit secondary antibody (1:100 in 3% BSA) conjugated to Alexa Fluor 488 (Molecular Probes, Inc.; Eugene, OR) for 30 min at 37C and then washed before viewing. For nestin immunocytochemistry, cells were fixed with 4% paraformaldehyde for 10 min, washed three times with 0.1 M PBS, and blocked with 2% normal goat serum (v/v) in 0.3% (v/v) Triton X-100 in 0.1 M PBS for 1 hr at room temperature. Cells were incubated with 1:100 dilution of anti-nestin monoclonal antibody (BD Biosciences Pharmingen; Mississauga, Ontario, Canada) overnight at 4C, washed for 30 min with 0.1 M PBS, and then incubated with goat anti-mouse secondary antibody conjugated to Alexa Fluor 568 (Molecular Probes Inc.) for 1 hr at room temperature. Negative controls were obtained by omission of the primary antibody.
For immunohistochemistry, frozen sections were thawed, rehydrated in 0.1 M PBS, and blocked with 3% (v/v) normal goat serum in 0.1% (w/v) BSA in 0.1 M PBS for 1 hr. Sections were incubated with anti-GFP (1:500) overnight at 4C, washed for 30 min, and then incubated with goat anti-rabbit Alexa 488 (1:500) for 1 hr. For nestin immunohistochemistry alone or following anti-GFP immunostaining, sections were blocked with 2% (v/v) normal goat serum in 0.3% (v/v) Triton X-100 in 0.1 M PBS for 1 hr, incubated with anti-nestin monoclonal antibody (BD Biosciences Pharmingen) overnight at 4C, washed for 30 min, and then incubated with goat anti-mouse secondary antibody conjugated to Alexa Fluor 568 for 1 hr. For immunostaining with cell type-specific markers, sections were blocked as described above and then incubated overnight at 4C with one of the following monoclonal antibodies: anti-glial fibrillary acidic protein (GFAP) 1:200 to detect astrocytes (Chemicon; Temecula, CA), anti-CC1/APC 1:1000 to detect oligodendrocytes (Calbiochem; San Diego, CA), and anti-microtubule-associated protein (MAP-2) 1:500 to detect neurons (Chemicon). After washing in 0.1 M PBS, sections were incubated for 1 hr with 1:500 goat anti-mouse Alexa 568. Negative controls were obtained by omission of the primary antibody, and for GFP controls, tissue sections were obtained from wild-type SD rats.
Results
GFP Expression in Adult CNS Tissues of Transgenic Rodent Strains
Tissue sections obtained from both neural and nonneural organs from the transgenic adult rodent strains were examined for native GFP expression with direct fluorescence under 488-nm excitation light. Adjacent sections were processed for GFP immunostaining to enhance native GFP fluorescence. Representative images of native GFP expression and immunostained tissue sections were obtained under identical imaging conditions. In all tissues, GFP immunohistochemistry resulted in increased GFP signal relative to direct fluorescent visualization of native GFP expression (shown only for the rat strains in Figure 1). Nonspecific cross-reactivity with the anti-GFP antibody was not observed in either transgenic or native tissue with the immunostaining conditions used in this study. Sections were counterstained with the nuclear dye DAPI to show tissue morphology, and a representative DAPI-stained image from the SD strain is shown for all the tissues. We compared GFP expression in the rat strains to a constitutively expressing mouse strain, because GFP mice are widely used and provide a good basis for comparison of expression patterns. In the study that generated the transgenic W rat strain (Hakamata et al. 2001), tissues were examined for GFP expression by fluorescence imaging of whole organs. In the present study, we examined GFP expression in tissue cryosections by both direct fluorescence and GFP immunostaining and found tissue- and cell type—specific expression patterns. Hadjantonakis et al. (1998) reported on the native expression of GFP in certain tissues of the M strain, but no reports have examined GFP fluorescence in tissues derived from the SD rat strain. We observed variations in GFP fluorescence intensity in the same tissue derived from different animals of the SD strain, and the most representative sections are shown. This variation in expression was not seen with the other transgenic strains.
In the spinal cord, very little GFP expression was evident in the SD rat strain even with immunohistochemical staining (Figures 1B and 1C). In the W strain, ventral horn motor neurons expressed GFP (Figures 1D and 1E), and in the transgenic mouse (M), GFP was strongly expressed throughout the vasculature, but minimally in the neurons and glia (Figure 1F). In both the SD and W transgenic rat strains, GFP was only weakly expressed in the central canal (Figures 1B-1E, insets), whereas in the M strain, some ependymal cells lining the central canal expressed GFP (Figure 1F, inset).
In the retina, GFP was weakly expressed in the ganglion cell layer (GCL) and inner nuclear layer (INL) of the SD transgenic rat (Figures 1H and 1I). In comparison, the W strain showed strong GFP expression in the GCL and the outer nuclear layer containing the photoreceptor cells (Figures 1J and 1K). In the M transgenic, GFP appeared to be expressed by a subset of neurons in the INL in addition to expression in the GCL (Figure 1L).
Whole sagittal brain sections were examined for native GFP expression; adjacent sections were processed for anti-GFP immunohistochemistry, but only the cerebellum is shown. In general, there was much lower GFP expression apparent in brain sections of the SD transgenic relative to the W strain (data not shown). In the cerebellum, very low levels of diffuse GFP expression were apparent in the SD transgenic (Figures 1N and 1O). In contrast, GFP was most strongly expressed by Purkinje neurons in the Purkinje cell layer of the W strain (Figure 1P), which was even more apparent in the adjacent section processed for GFP immunohistochemistry (Figure 1Q). In the M strain, GFP was expressed in the molecular layer and the vasculature of the cerebellum (Figure 1R). GFP expression was also examined in other brain regions, summarized in Table 1. In addition to the ependymal region of the spinal cord, adult stem/progenitor cells have also been reported in the subventricular zone of the lateral ventricle and subgranular zone of the dentate gyrus. However, there was no detectable GFP expression in these regions in either the SD or W rat strains. In contrast, the M strain showed strong GFP expression in the lateral ventricle/subventricular zone and weak expression in the hippocampus and dentate gyrus (see Table 1). These results suggest that GFP expression is not a uniform property of all neural stem cells isolated from a certain transgenic strain.
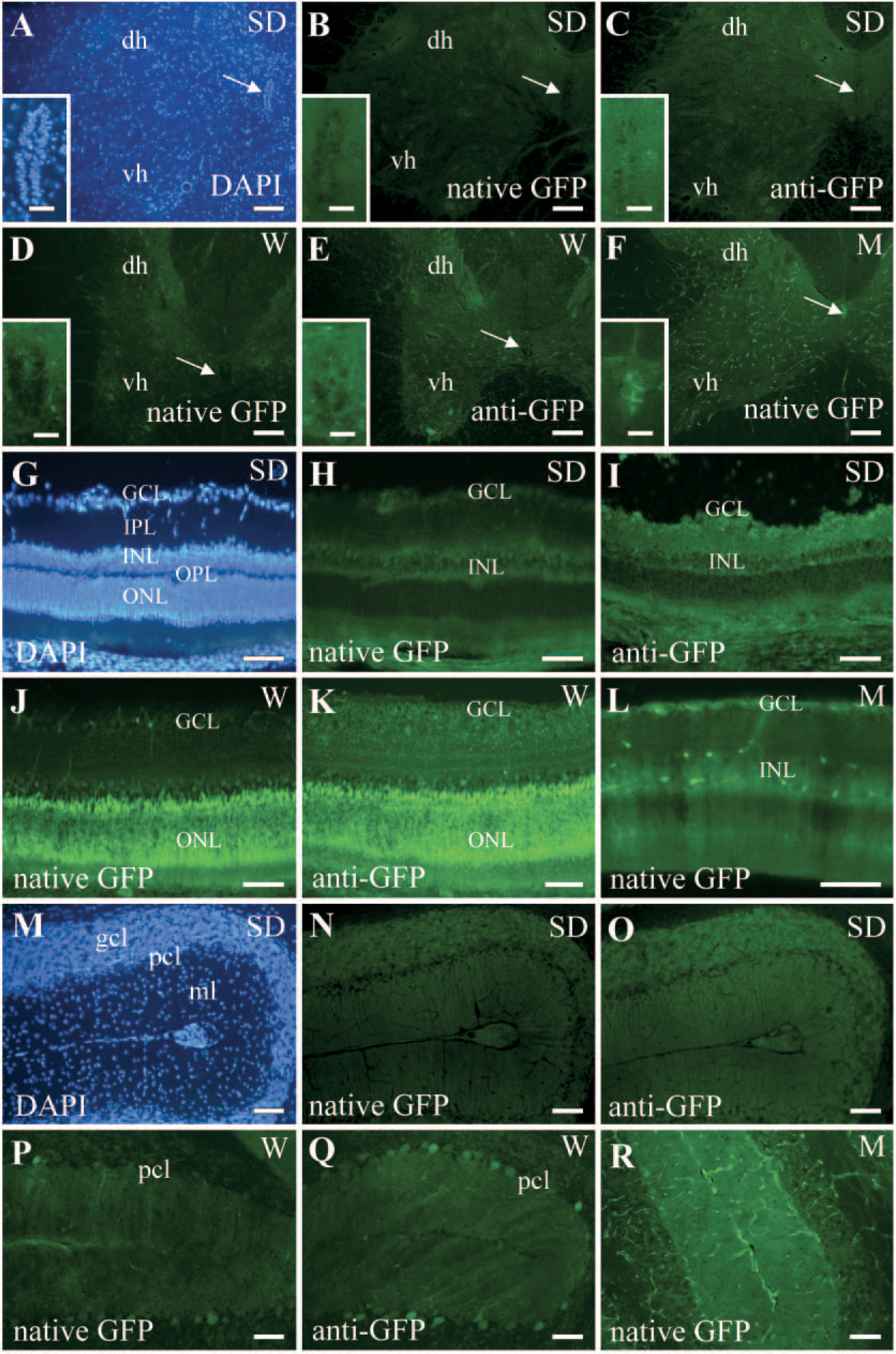
Green fluorescent protein (GFP) expression in the central nervous system by direct fluorescence and immunohistochemistry. Native GFP expression was visualized directly by fluorescence imaging (native GFP as indicated in the panels) and immunohistochemistry, with the anti-GFP antibody performed on adjacent sections (panels labeled anti-GFP).
Differential native green fluorescent protein expression in neural and non-neural tissues examined in adult transgenic rodent strains
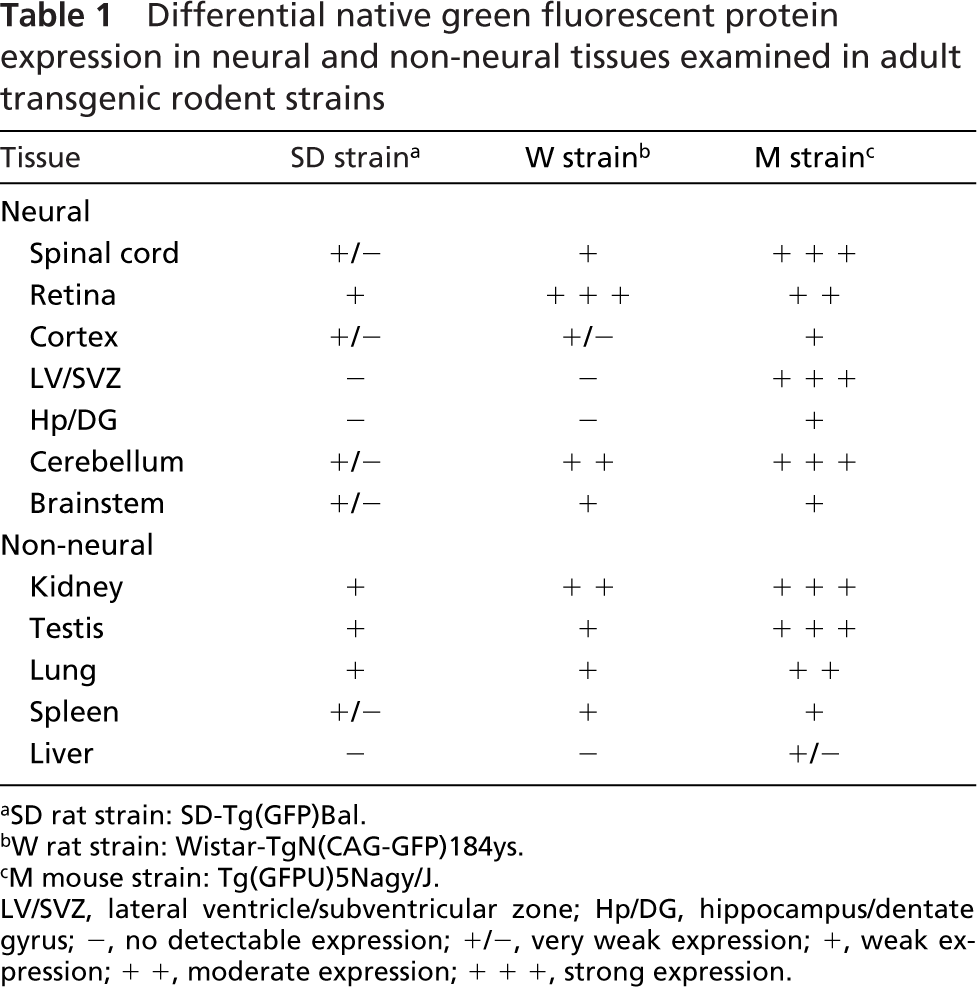
SD rat strain: SD-Tg(GFP)Bal.
W rat strain: Wistar-TgN(CAG-GFP)184ys.
M mouse strain: Tg(GFPU)5Nagy/J.
LV/SVZ, lateral ventricle/subventricular zone; Hp/DG, hippocampus/dentate gyrus; -, no detectable expression; +/-, very weak expression; +, weak expression; + +, moderate expression; + + +, strong expression.
GFP Expression in Adult Non-neural Organs
Non-neural organs (shown are kidney and testis, Figure 2) were also examined for GFP expression with direct fluorescence and immunohistochemical analysis for comparison with the neural expression pattern. Results shown in Figure 2 are all native GFP fluorescence. Similar to the neural tissues (Figure 1), GFP immunostaining of sections of non-neural tissue also resulted in a more intense GFP signal when compared with the native fluorescent signal (data not shown).
In the SD strain, sections of kidney cortex showed weak expression of GFP in the convoluted tubules (Figure 2B, arrows), whereas GFP was strongly expressed in glomeruli (Figure 2C, arrowheads) in the W transgenic. High levels of GFP expression were apparent in glomeruli (Figure 2D, arrowheads) and tubules (Figure 2D, arrows) in the M transgenic, as seen previously (Hadjantonakis et al. 1998). Transverse sections of testis were also examined, and as shown in Figures 2F and 2G, diffuse expression of GFP was observed in the seminiferous tubules in both the SD and W strains. In the M strain, strong GFP fluorescence was also apparent around the tubules (Figure 2H). The lung and spleen also expressed GFP in all three transgenic rodents (data not shown) as demonstrated previously for the M and W strains (Hadjantonakis et al. 1998; Hakamata et al. 2001), and GFP expression was not detected in liver tissue in the SD and W strains (data not shown, summarized in Table 1).
Neurospheres Derived from the Ependymal Region of Adult GFP Rat Spinal Cord
In culture, neural stem/progenitor cells grow in suspension in defined medium supplemented with mitogens as spherical aggregates called neurospheres (Gritti et al. 1996; Weiss et al. 1996). Neurospheres consist mainly of progenitor cells that are more restricted in their proliferative and phenotypic potential and a small number of multipotent stem cells.
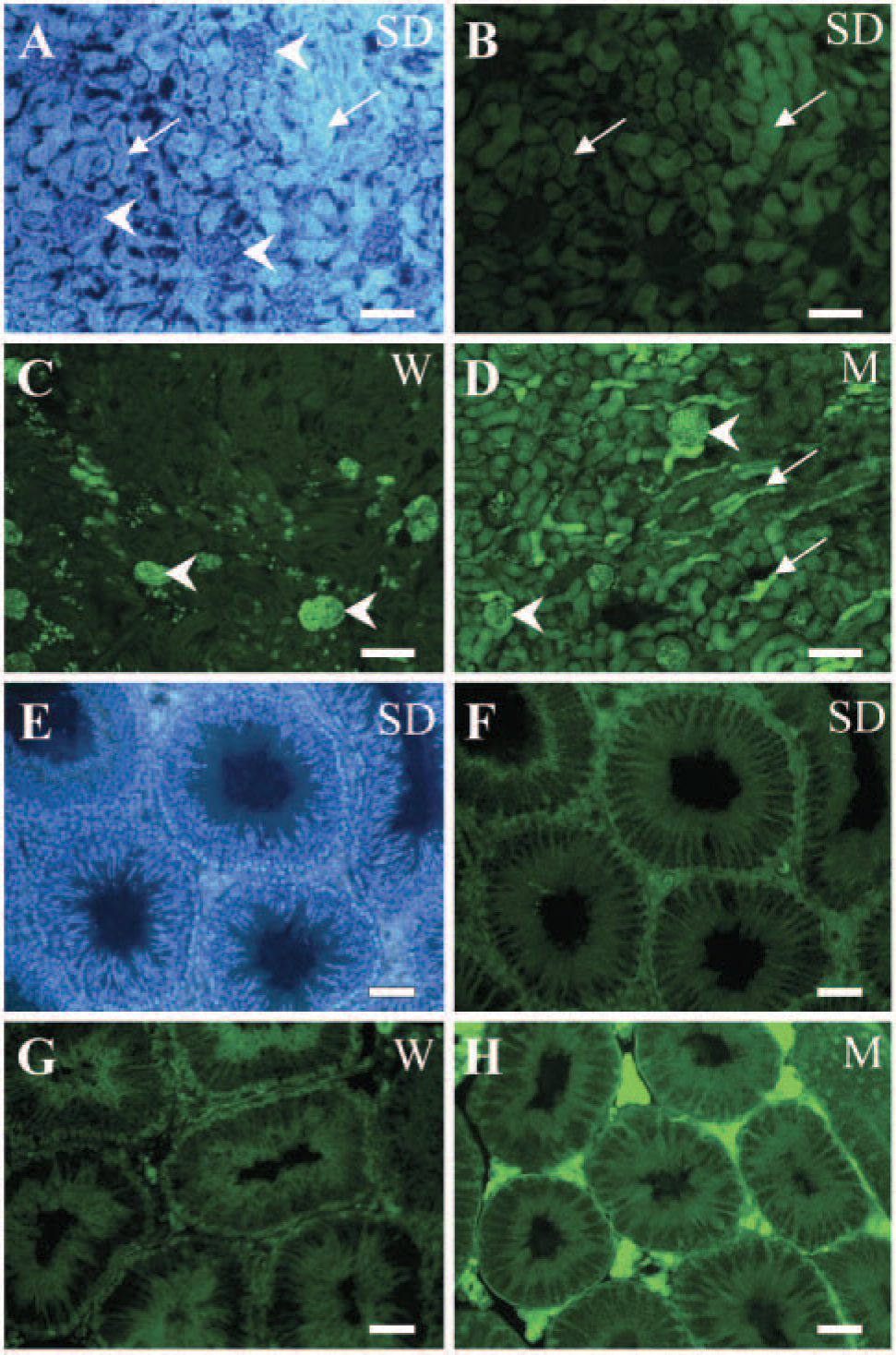
GFP expression patterns in non-neural tissues in transgenic rodents. Native GFP expression was visualized by direct fluorescence in the kidney
Neurospheres were generated from the ependymal region of the adult spinal cord from either the SD or W rat strains. Floating neurospheres were observed by day 5 in vitro, and GFP fluorescence was examined immediately after initial culture and at passages 1 to 4 thereafter. Neurospheres generated from the SD strain showed barely detectable native GFP expression (Figure 3B). However, when the neurosphere cultures were immunocytochemically stained with the anti-GFP antibody, strong fluorescent GFP signal became apparent (Figure 3D). In contrast, neurospheres generated from the W strain showed strong native GFP expression in culture (Figure 3F) without the need for immunocytochemical processing. Native expression of GFP was retained after dissociation of the neurospheres into single cells (Figure 3F, inset). As shown in Figure 3H, neurospheres derived from the spinal cord of the transgenic rats also showed nestin immunoreactivity, which is a marker of stem/progenitor cells in the CNS (Lendahl et al. 1990). In addition, we have determined that these neurospheres generated from the adult spinal cord show properties of stem cells, because they are self-renewing, they can be propagated in vitro, and they are clonally derived and multipotential (Kulbatski et al., unpublished data). When the neurospheres were plated onto an adherent substrate and mitogens were withdrawn and bovine serum was added, cells migrated from the sphere and differentiated into neurons, astrocytes, and oligodendrocytes, as identified with cell type—specific antibodies (data not shown).
Transplantation of GFP-expressing Spinal Cord Stem/Progenitor Cells into Wild-type Spinal Cord
Neurospheres derived from the adult spinal cord of the SD or W rat strain were dissociated, and these stem/progenitor cells were transplanted into the intact spinal cord of wild-type rats to assess the ability to track the transplanted cells in vivo. Spinal cord stem/progenitors generated from the SD transgenic strain were detected after transplantation with GFP immunohistochemistry (Figure 4A). When viewed with direct fluorescence, native GFP expression was not detectable in the transplanted cells, as was the case in culture (Figure 3B). In comparison, robust native expression of GFP was observed by direct fluorescence when the transplanted cells were derived from the W strain (Figure 4D). Strong native expression of GFP in transplanted cells derived from the W transgenic was still evident at 49 days after transplantation (Figure 4G). Therefore, GFP-positive grafted cells survived at least 49 days after transplantation (Figure 4G), and had migrated from the site of injection (data not shown). Also, at 3 days after transplantation, the grafted stem/progenitor cells expressed nestin (Figures 4B, 4C, 4E, 4F, arrows). By 49 days following transplantation, nestin was no longer expressed by the transplanted cells that continued to show strong native GFP fluorescence (Figures 4G-4I) and were now expressing primarily mature glial phenotypes (as shown in Figure 5). Nestin was still upregulated in the spinal cord around the grafting site at 49 days after transplantation (Figure 4H) because of nestin expression by reactive astrocytes, as has been shown following a lesion (Frisen et al. 1995). GFP-expressing grafted cells differentiated into astrocytes, as detected by the GFAP antibody (Figures 5A-5C), and oligodendrocytes, as detected by the CC1/APC antibody (Figures 5D-5F). Transplanted cells did not differentiate into neurons, as shown by the lack of MAP-2 colocalization (Figures 5G-5I).
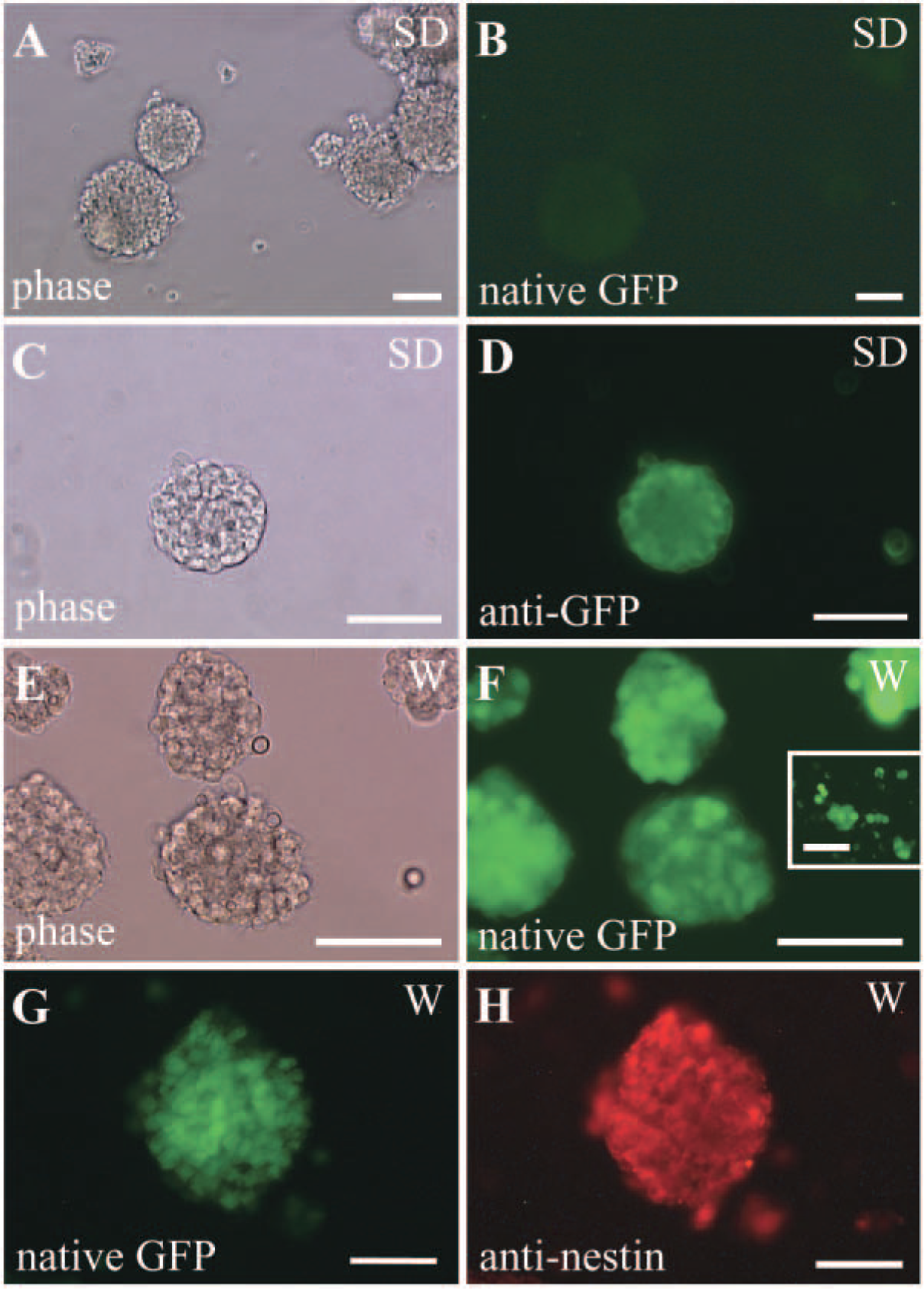
Stem/progenitor cells cultured as neurospheres from the spinal cord ependymal region of GFP transgenic rats.
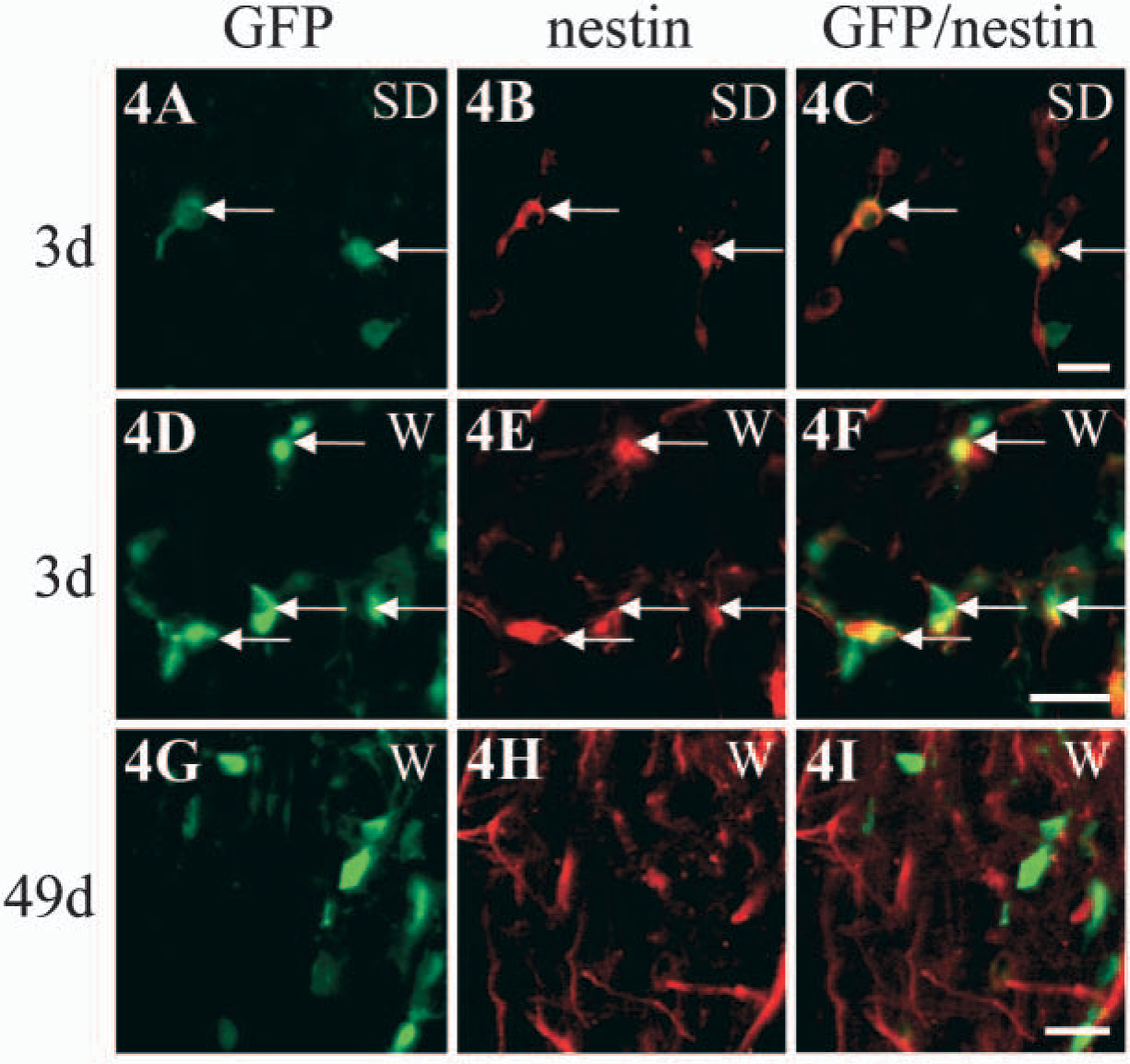
Transplantation into the spinal cord in vivo of ependymal region stem/progenitor cells derived from GFP transgenic rat spinal cord.
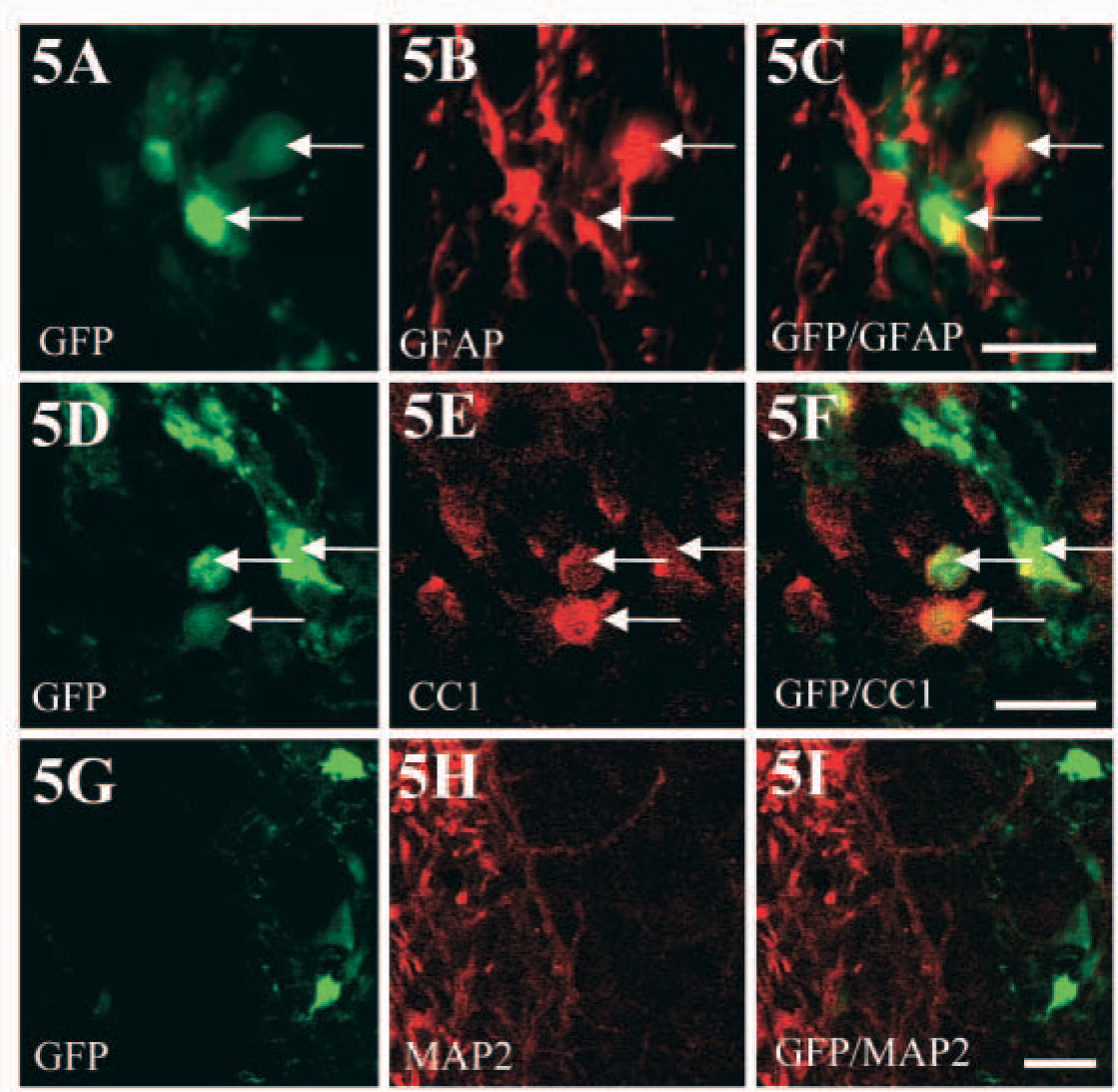
Phenotype of grafted stem/progenitor cells derived from the W strain at 49 days after transplantation into the spinal cord.
Discussion
A number of different methods can be used to label cells for subsequent transplantation studies including pre-labeling in vitro with nuclear labels such as bromodeoxyuridine, cytoplasmic labels such as carboxy-fluorescein diacetate, and membrane labels such as DiI (Onifer et al. 1993). However, one of the many disadvantages to utilizing these markers for labeling transplanted cells is that when cells divide, either in culture or in vivo, the label is progressively diluted. Reporter gene expression does not undergo dilution as a result of cell proliferation, so it is suitable for long-term transplantation studies. Another important advantage of reporter genes is that they can be stably integrated into the genome and will not transfer to host cells (Cao et al. 2002). Cells can be pre-labeled in vitro with reporter genes cloned into a variety of vectors where detection of the reporter gene product is dependent on the stable and sustained expression of the transgene in transplanted cells. Unfortunately, a major disadvantage of transfection of cells in culture with viral vectors is that reporter gene expression can be significantly downregulated over time in transplanted cells (Onifer et al. 1993; Vroemen et al. 2003). However, a more sustained expression of the transgene in grafted cells in vivo is obtained when transplanted cells are derived from transgenic animals.
A variety of reporter genes have been used in transgenic animals, the most common of which are lacZ, which encodes the Escherichia coli enzyme β-galactosidase, human placental alkaline phosphatase (hPAP), and GFP. Transgenic rats containing the lacZ and hPAP reporter genes have also been developed (Kisseberth et al. 1999; Takahashi et al. 2003). Transplanted cells derived from the lacZ rat strain have been tracked in vivo following myocardial injury and in a cerebral infarction model (Takahashi et al. 2003; Inoue et al. 2005). Neuronal-restricted precursors derived from the hPAP transgenic rat (Kisseberth et al. 1999) were also shown to consistently mark cells following intraspinal grafting (Mujtaba et al. 2002). The visualization of these enzyme-based reporters requires fixation of the sample and a chromogenic enzyme-based reaction. Fluorescent protein reporters such as GFP are advantageous because GFP fluorescence can be visualized directly, without further processing (Hadjantonakis and Nagy 2001). The cloning of GFP was the first example of a gene-based fluorescent protein reporter that is intrinsically fluorescent (Prasher et al. 1992). There are a number of advantages to utilizing GFP for tracking transplanted cells, including that it is constitutively fluorescent and so is readily detected when under the control of a constitutive promoter, it has a high fluorescence yield, and it is resistant to photobleaching. In addition, GFP has no endogenous counterpart in mammals, ensuring specificity, the signal can be amplified immunohistochemically, the signal can be detected in both cultured and fixed cells, and GFP-expressing cells can be enriched or sorted with other markers using fluorescence-activated cell sorting prior to transplantation.
A number of methods can be used to introduce the reporter gene, including pronuclear injection, retroviral-mediated gene transfer, or gene transfer in embryonic stem cells. The classic and most widely used route for the production of transgenics is through the introduction of a DNA construct linking a promoter/enhancer element to the reporter gene into zygotes by pronuclear injection (Hadjantonakis and Nagy 2001). The W rat strain examined in the present study was generated by pronuclear injection. Transgenic rodents made through pronuclear injection have the advantage that the injected DNA integrates into the genome in tandem copies, so that very high levels of expression can be achieved (Tucker 2001). This method allows the production of large numbers of transgenics, each achieved through germline transmission of the founder animal's genome. However, this is an expensive and time-consuming process, and the production of transgenics by microinjection has low efficiency. Transgenic animals can also be generated by the transfection of embryonic stem cells, which are subsequently transplanted to blastocysts; the M strain used in the present study was generated in this way. The advantage of using embryonic stem cells over DNA injection is that the genome can be manipulated through homologous recombination and the transgenic cell line can also be propagated and studied (Tucker 2001). However, as with all embryonic stem cell-mediated approaches, establishment of a transgenic line requires colonization of the germline of a chimera (Hadjantonakis and Nagy 2001). Transgenics can also be generated by retroviral-mediated gene transfer. Lentiviral vectors are the most commonly used, because they are able to transduce both dividing and non-dividing cells. Lentiviral-mediated gene transfer was used to generate the SD strain examined in the present study. Virally delivered transgenes efficiently integrate into the genome, and stable and long-term expression of the GFP gene was observed in transgenic mice generated by lentiviral vectors (Pfeifer et al. 2002; Ikawa et al. 2003). The disadvantages with lentiviral-mediated gene delivery include the potential for viral toxicity and random integration of the transgene into the genome, which makes it difficult to establish pure-breeding transgenic lines (Ikawa et al. 2003).
The GFP transgenic rat strains used in this study have a number of differences. First, these rats differ in background strain, SD or Wistar. Second, different constitutive promoters drive the GFP gene. The SD strain contains the ubiquitin-C promoter, whereas the W strain contains the chicken β-actin promoter. These differences can result in tissue-specific variation in GFP fluorescence intensity, as we have observed in both neural and non-neural tissues. Also, these strains were generated differently, as discussed above, so there are insertion site differences of the GFP transgene. As noted above, the SD strain was generated via lentiviral transfection, whereas the W strain was generated by pronuclear injection. A transgene delivered by retroviral vectors usually integrates as a single provirus into multiple loci, whereas pronuclear injection often results in an integration of multiple copies into a single locus (Nakanishi et al. 2002). Thus, the random integration of the transgene in the SD strain can result in the variation of GFP expression observed in individual rats. In our examination of GFP expression patterns in neural and non-neural tissues of the SD strain, we observed some variation of fluorescence intensity between individual rats. Unfortunately, this is a drawback to the use of the SD strain for generating cells for transplantation, because comparable GFP expression levels from different animals are required for consistency in fluorescence intensity. Recently, however, this SD strain was further developed by the RRRC supplier, and the new SD line was genetically stabilized so that the transgene integrated to a single insertion site, thereby eliminating the variability in expression levels we have seen with the earlier SD strain used in the present study. Also, recently the transgene construct in the W strain was developed in an inbred Lewis rat strain (Inoue et al. 2005). This new Lewis strain expresses GFP strongly and ubiquitously in most of the organs, compared with the Wistar GFP transgenic line used in the present study.
When native GFP expression in various neural and non-neural tissues of the transgenic strains was compared with the signal obtained with immunostaining, a more intense GFP signal was observed in tissues processed for immunohistochemistry. In the region of the central canal in the spinal cord of both the SD and W transgenic rats, very low levels of GFP expression were apparent in vivo, even with immunohistochemical processing. When cells were cultured from the region of the central canal and passaged as neurospheres, spheres generated from the SD strain showed barely detectable native GFP fluorescence in vitro, consistent with the in vivo expression pattern. In contrast, cultured cells generated from the W strain showed robust native GFP fluorescence in vitro, but in vivo there was only a low level of GFP fluorescence. It is possible that the low levels of GFP expression observed in cells in the region of the central canal of the W transgenic were below our level of detection because of incomplete penetration of the anti-GFP antibody. We did not investigate alternative methods of optimizing immunostaining conditions, so we cannot exclude this possibility. The fluorescence intensity of GFP is dependent on a number of factors, including protein folding, dimerization status, temperature, pH, and oxidative conditions (Tsien 1998), so high amounts of a protein do not necessarily mean that this protein also produces fluorescent signaling (Zhang et al. 2001). Also, it is possible that the culture conditions induced the expression of GFP in cells derived from the W transgenic because the transgene is driven by different promoters in the two strains.
Stem/progenitor cells cultured from either the SD or W transgenic strains were identified in vivo following transplantation into the wild-type spinal cord, although anti-GFP immunohistochemistry was required to visualize grafted cells generated from the SD strain. Also, the fluorescent GFP signal of grafted cells observed at 3 days following transplantation was more robust in grafted cells derived from the W strain. Neurospheres in culture express nestin, a marker for stem/progenitor cells. When spheres are differentiated, nestin expression is downregulated as phenotypes of mature cells are expressed. Likewise, in vivo at 3 days after transplantation, grafted cells expressed nestin, which was then downregulated by 49 days after transplantation. By this time, transplanted cells had differentiated into astrocytes and oligodendrocytes, as shown by cell type-specific markers. The phenotype of the transplanted GFP-expressing cells was evaluated with cell type-specific antibodies within each cell lineage to show that the transgene marker is useful for tracking all cell types within a transplant. These data show that cultured cells generated from GFP-expressing rats can be easily visualized and tracked in vivo after transplantation into the spinal cord with direct fluorescence or anti-GFP immunohistochemistry. Also, low levels of GFP expression visualized in vivo in certain tissues do not rule out the utility of culturing cells from these tissues and utilizing them for transplantation studies, because, as was shown here, GFP fluorescence could be enhanced by culture conditions or by anti-GFP immunocytochemistry.
Several strains of transgenic mice expressing GFP under the control of constitutive, cell-specific, or inducible promoters have been established by either classic pronuclear injection (Okabe et al. 1997; Young et al. 1999; Fischer et al. 2000; Yamaguchi et al. 2000; Zhang et al. 2001; Mignone et al. 2004), embryonic stem cell-mediated transgenesis (Hadjantonakis et al. 1998), or lentiviral vector-mediated transgenesis (Lois et al. 2002; Pfeifer et al. 2002; Ikawa et al. 2003). Although it has been more difficult to generate transgenic rats, some of the GFP strains available have been developed by pronuclear injection (Hakamata et al. 2001) and lentiviral vector delivery (Lois et al. 2002). The advantage of generating cells from GFP-expressing rats for transplantation into rat hosts is that it is technically simpler to perform transplants in rats than in mice for studies such as spinal cord regeneration, and this allows for syngenic transplant studies without the potential concerns of donor cell rejection.
We have shown that utilizing the SD or W transgenic rat strains that constitutively express GFP as a marker for identifying transplanted cells in situ has a number of distinct advantages, including the specificity of fluorescence signal and the stable and retained expression of GFP in vivo for long-term studies. We have shown that GFP-expressing stem/progenitor cells derived from the spinal ependymal region can be detected for at least 49 days after transplantation into the spinal cord and that these differentiate into astrocytes and oligodendrocytes. However, we cannot exclude that there is partial silencing of the transgene after transplantation. We have also determined that generating cells for transplantation from the GFP-expressing W rat strain is advantageous compared with the SD strain, because GFP expression of cells derived from the W rat strain is readily detected by direct fluorescence both in vitro and in vivo, without the need for immunohistochemical amplification of the GFP signal. Also, GFP-expressing transplanted cells derived from the W strain can be readily identified, their migration can be tracked in situ, and their phenotype can be determined by immunostaining with cell type-specific antibodies.
Footnotes
Acknowledgements
This work was supported by grants from the Ontario Neurotrauma Foundation, Canadian Paraplegic Association (Ontario Branch), and Physicians’ Services Incorporated (CHT), and by the Gloria and Seymour Epstein Chair in Cell Therapy and Transplantation (AK). A.J.M. was supported by an Ontario Neurotrauma Fellowship.
We wish to thank Huijie Jiang for maintaining a steady supply of Wistar-TgN(CAG-GFP)184ys rats for these studies (originally generated by Dr. E. Kobayashi). We would also like to thank Dr. B. Bauer of the Rat Resource and Research Center, University of Missouri, Columbia, MO, for her very helpful comments.