
Abstract
This study demonstrated determination of fetal gender from nucleated red blood cells (NRBCs) in maternal blood and attempted to apply prenatal diagnosis of hemophilia A using BclI DNA polymorphism. Venous blood was drawn from 20 pregnant women, and NRBCs were recovered by magnetic activated cell sorting and anti-GPA (glycophorin A) immunostaining. After microdissector isolation of the NRBCs, primer extension preamplification (PEP) and nested PCR of the amelogenin gene were performed to determine fetal gender. We also performed PEP and nested PCR of BclI polymorphism to verify the validity of prenatal diagnosis of hemophilia A. DNA amplification was achieved in 107 cells (51.9%) and fetal gender determined with 65.0% accuracy. Unfortunately, we could not verify the validity within the scope of this study. However, in a larger number of cases that are informative in BclI polymorphism, we will be able to identify patients affected by hemophilia A using fetal NRBCs in maternal blood.
Keywords
C
Various immunological traits have been tried to enrich fetal NRBCs. We used magnetic activated cell sorting (MACS) for CD71 (transferrin receptor) of NRBCs. After positive MACS with anti-CD71 antibody, we could expect that the magnetically labeled cell suspension eluted from the MACS column contains NRBCs, activated lymphocytes, and macrophages. Because cells of erythroid lineage have glycophorin A (GPA) as a cell membrane antigen, we used immunostaining with an anti-GPA antibody to differentiate fetal NRBCs in cell suspension, which eluted after MACS for CD71. It has been well established that this combination of antigens is excellent for isolation of nucleated erythrocytes (Bianchi et al. 1990,1992; Price et al. 1991; Wachtel et al. 1991; Ganshirt-Ahlert et al. 1992; Reading et al. 1995). However, even after separation with monoclonal antibodies, maternal cell contamination is unavoidable. Therefore, we picked up a single cell with a micromanipulator (Takabayashi et al. 1995).
The problem is that NRBCs in maternal peripheral blood are of both fetal and maternal origin (Ganshirt et al. 1994; Slunga-Tallberg et al. 1995,1996; von Eggeling et al. 1997; Holzgreve et al. 1998; Troeger et al. 1999). However, despite the fact that nucleated erythrocytes in maternal peripheral blood are of fetal and maternal origin, many researchers have claimed that they can diagnose various disorders using NRBCs in maternal peripheral blood, such as hemoglobinopathies (Di Naro et al. 2000), Rhesus type, and Duchenne muscular dystrophy (Sekizawa et al. 1996a,b).
Therefore, we focused on the possibility of diagnosis of hemophilia A using fetal nucleated erythrocytes in maternal peripheral blood. In Korea, PCR-based analysis of the BclI/intron 18 and St14 VNTR polymorphisms is useful in carrier detection and prenatal diagnosis of hemophilia A, and the observed heterozygosity for BclI/intron 18 polymorphism was 21.0% (Choi et al. 2000). Because it is very difficult to apply St14 VNTR analysis in single-cell PCR, we tried BclI polymorphism to diagnose hemophilia A.
In this study, we first tried to examine the efficacy of a noninvasive method by using fetal NRBCs in maternal peripheral blood to determine fetal gender. Second, we aimed to check the validity to diagnose hemophilia A by BclI polymorphism using fetal cells in maternal blood.
Materials and Methods
Peripheral blood samples (20 ml) were collected in EDTA tubes from 20 healthy women with uncomplicated singleton pregnancies (10 male, 10 female fetuses) between 10 and 22 weeks of gestation. This patient population included nine primigravidas and 11 multigravidas referred to our antenatal testing unit for amniocentesis or chorionic villus sampling (CVS) because of advanced maternal age and abnormal triple screen results. All patients were Rh positive. The samples were collected before amniocentesis or CVS was performed.
Isolation and Enrichment of Nucleated Cells
The blood samples were diluted with a double volume of PBS. Nucleated cells were isolated by density gradient centrifugation using Ficoll-Paque (Pharmacia Biotech; Uppsala, Sweden). Centrifugation was performed at 400 × g for 30 min. The layer of cells between plasma and Ficoll-Paque, in which most mononucleated cells were recovered, was washed with PBS, pH 7.2, and centrifuged at 300 × g for 10 min. After the washing procedure was repeated and the supernatant decanted, the cell pellet was resuspended in PBS to allow counting of the total number of recovered cells.
After centrifugation and resuspension of the pellet in PBS [supplemented with 0.5% bovine serum albumin (BSA) and 2 mM EDTA], the samples were incubated for 15 min at 4C with a 1:10 solution of monoclonal antibodies (CD 71; Miltenyi Biotec, Bergisch Gladbach, Germany) and inactivated plasma. Separation was achieved with the mini-MACS system (Miltenyi Biotec) according to the manufacturer's recommendations.
Slide Preparation and Immunophenotyping
After mini-MACS separation, in theory the eluted cell suspension would contain NRBCs, activated lymphocytes, and macrophages. We tried to separate NRBCs from the remaining cells using anti-GPA antibody. Therefore, we made slides for microscopic examination by cytocentrifugation (Shandon Southern; Runcorn, U.K.). The slides were fixed in cold acetone for 5 min. Anti-GPA (mouse anti-human glycophorin A) monoclonal antibody (DAKO; Carpinteria, CA) was used to identify NRBCs by 3-amino-9-ethyl-carbazole immunostaining (DAKO). Hematoxylin counterstaining was used for morphological characterization of the cells. Finally, the immunophenotyped cells were classified under a light microscope. The coordinates of GPA-positive nucleated erythrocytes on the slides were recorded and the cells photographed.
Single-cell Isolation by Microdissector and Fine Needle
Single NRBCs, recognized as red by anti-GPA immunocytochemistry, were collected with a 30-G needle and microdissector (Figure 1). Each NRBC was transferred to its own tube containing 5 μl distilled water and then snap-frozen in liquid nitrogen. Protein digestion was carried out for 2 hr at 60C by adding 5 μl proteinase K (500 μg/ml)/buffer mixture (10 mM Tris-HCl, pH 8.3, 50 mM KCl). After complete digestion, proteinase K was inactivated by heating to 94C for 2 min.
Amplification of DNA by Primer Extension Preamplification
Primer extension preamplification (PEP) was performed as described by Zhang et al. (1992). Briefly, the initial reaction volume containing the digested single cell was brought to a final volume of 60 μl with 33 μM totally degenerated 15-mer primer (Operon Technologies; Alameda, CA), PCR buffer containing 10 mM Tris-HCl, pH 8.3, 50 mM KCl, 2.5 mM MgCl2, 100 μM dNTPs, and 5 U Taq polymerase (Takara; Tokyo, Japan). Fifty cycles were carried out in a PTC 200 thermocycler (MJ Research; Cambridge, MA). Each cycle consisted of 1 min denaturation at 92C, 2 min annealing at 37C, heating to 55C at 0.1C/sec, and 4 min extension at 55C. After PEP, the PCR product was stored at 4C as a template for nested PCR.
Nested PCR of Amelogenin Loci and BclI Polymorphic Loci
We used nested PCR amplification of the amelogenin gene on chromosomes X and Y for gender determination of the samples (Schaaff et al. 1996). The following procedure was used for PCR: 45 μl of PCR mix was added to each 5 μl PEP aliquot, resulting in a total reaction volume of 50 μl and final concentrations of 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.001% (w/v) gelatin, 0.2 mM of each dNTP, 400 nM of each outer primer [AMG 3 (5'-CTT CCC AGT TTA AGC TCT GAT G-3'), AMG 4 (5'-CCT TGC TCA TAT TAT ACT TGA C-3')], and 1 U Taq polymerase. Samples were heated at 94C for 5 min to ensure DNA denaturation, followed by 30 cycles of denaturation at 94C for 30 sec, annealing at 52C for 60 sec, and extension at 72C for 60 sec. Final extension at 72C was allowed for 10 min. For nested amplification, 2 μl of amplified product was transferred to a new reaction tube containing 23 μl of fresh PCR mix. This mix was the same as outer PCR except that it contained 400 nM of each inner primer [AMG 5 (5'-CTC AGG GAG GTT CCA TGA-3'), AMG 6 (5'-TGA GAA AAC CAG GGT TCC-3')] in place of the outer primers, and the MgCl2 concentration was increased to 2.5 mM. The amplification cycle of inner PCR included denaturation at 94C for 30 sec, annealing at 56C for 30 sec, and extension at 72C for 60 sec. Final extension was performed at 72C for 10 min. Amplification products were electrophoresed on 2% agarose gel in 1 × tris borate EDTA with ethidium bromide and visualized using an ultraviolet transilluminator. A cell was considered to be genotypically female if one band of 323 base pairs (bp) was present and male if a band of 142 bp was also present.
After MACS and anti-GPA antibody immunostaining
BclI polymorphism (Snabes et al. 1994) was performed for seven pregnant women. The slightly modified PCR procedure was as follows. A 45-μl PCR mix was added to each 5-μl PEP aliquot, resulting in a total reaction volume of 50 μl and final concentrations of 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.25 μM of each dNTP, 0.25 mM of each outer primer [HA1F (5'-CTA CCT GGC TTA GTA ATG GCT C-3'), HA1R (5'-AAA GGA ATA AAT TCC TTT TCC C-3')], and 1.25 U Taq polymerase. Samples were heated at 92C for 3 min to ensure DNA denaturation, followed by 20 cycles of denaturation at 92C for 30 sec, annealing at 49C for 30 sec, and extension at 72C for 30 sec. Final extension at 72C was performed for 3 min. For nested amplification, 2 μl of amplified product was transferred to a new reaction tube containing 23 μl of fresh PCR mix. This mix contained 0.25 mM of each inner primer [HA2F (5'-ATC AAA GGA TTC GAT GGT ATC T-3'), HA2R (5'-TTT CCT TTT TAG CAA TTT TTC T-3')] in place of the outer primers. After initial denaturation at 92C for 3 min, 30 cycles of amplification were carried out for inner PCR (92C for 30 sec, 46C for 30 sec, and 72C for 30 sec for denaturation, annealing, and extension, respectively). Final extension was performed at 72C for 3 min. BclI (30 U) was added directly to the PCR product and after 1 hr at 50C, 10-μl aliquots were electrophoresed on a 2% agarose gel.
Results
The first part of this study aimed to examine the efficacy of a method using fetal cells in maternal peripheral blood by determining fetal gender using the amelogenin gene from 20 pregnant women.
In total, 206 cells were obtained from peripheral blood of 20 pregnant women. Among these, 107 cells (51.9%) could be amplified by PEP and nested PCR of the amelogenin gene.
In each of 20 cases, 1–25 NRBCs were screened with the amelogenin system for gender determination (Table 1). Gender was ascertained in each case by karyotyping of chorionic villi or amniocytes.
In this study, we assigned the fetus as male if we could find any single cell that showed a band corresponding to the Y-chromosome allele in amelogenin PCR. According to this rule, we correctly identified 8/10 male fetuses and 5/10 female fetuses. Combining these results, the accuracy of gender determination in our approach was 65.0%.
The second part of this study aimed to check the validity of diagnosing hemophilia A by BclI polymorphism using fetal NRBCs in maternal blood. We selected 85 NRBCs from seven pregnant women, of which 47 cells (55.3%) could be amplified by PEP/nested PCR of BclI polymorphism. However, only one of the seven pregnant women showed a difference between fetus (amniotic fluid) and mother (peripheral blood) in restriction fragment length polymorphism pattern. That is to say, BclI polymorphism was informative in only one pregnant woman. Because of lack of informative cases using BclI polymorphism in this study, we could not be sure whether prenatal diagnosis of hemophilia A by fetal NRBCs in maternal peripheral blood is applicable. However, in this case, in spite of a male-bearing pregnancy, three cells showed a female-specific band pattern in amelogenin PCR. In addition, these three cells showed the same band pattern as maternal cells in BclI polymorphism (Figure 2). Therefore, we could conclude that these three cells were of maternal origin.
Nucleated erythrocytes separated from 16 pregnant women amplified by PEP and amelogenin nested PCR
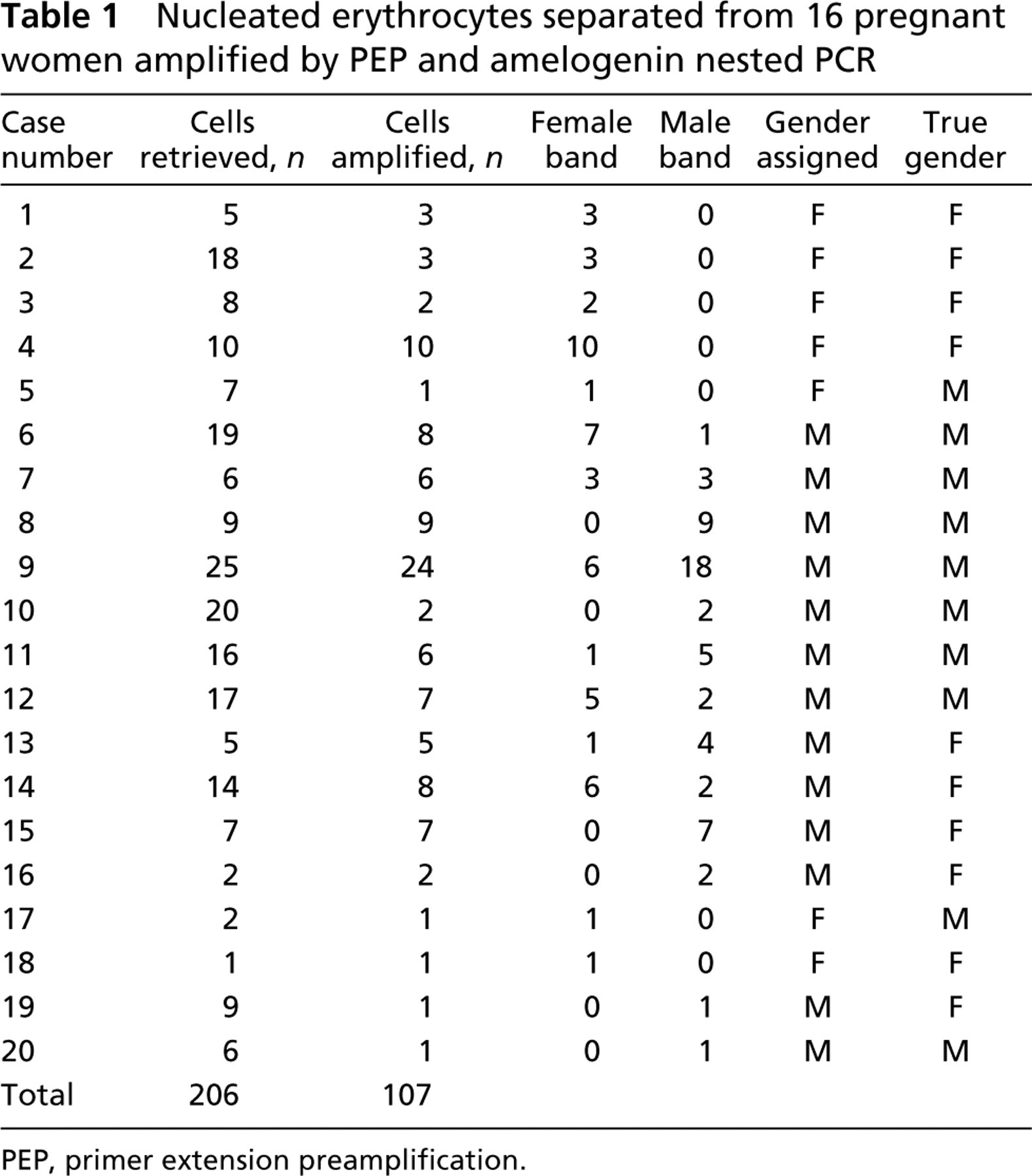
PEP, primer extension preamplification
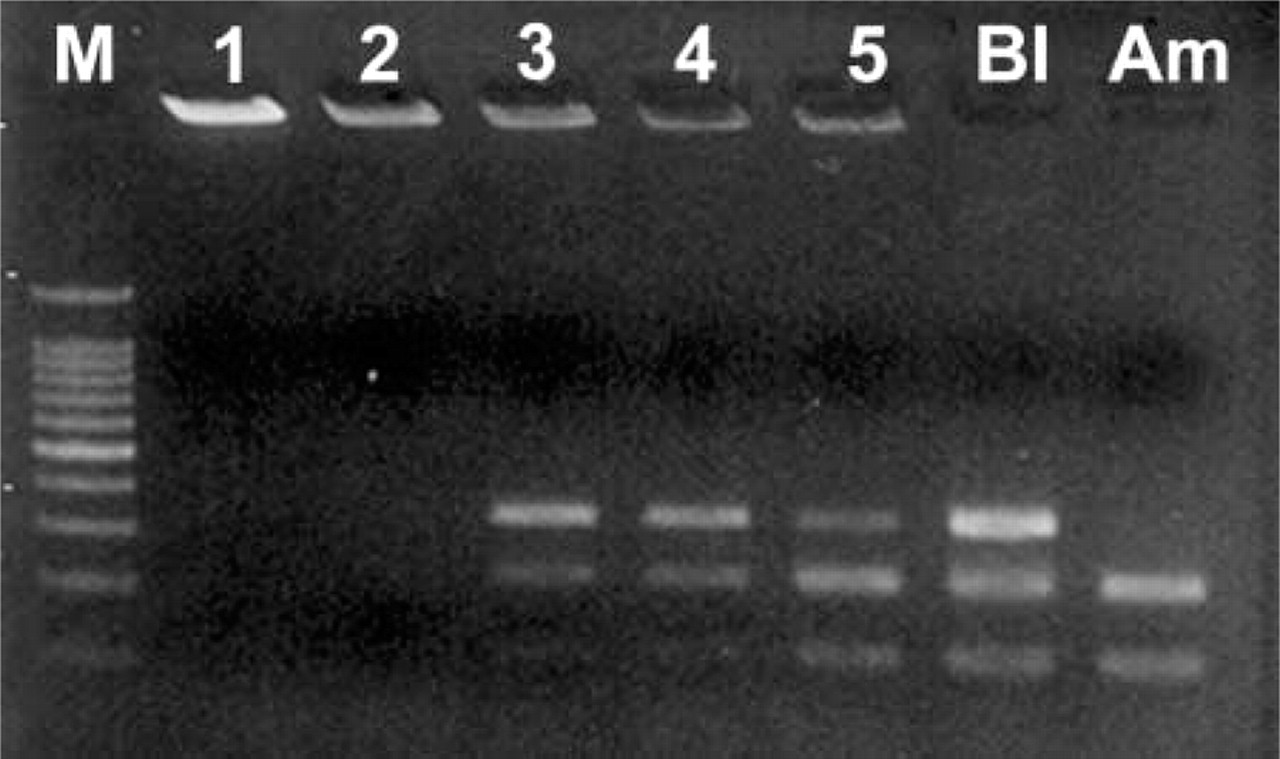
Agarose gel electrophoresis of BclI polymorphism results from a male fetus. Three cells (Lanes 3–5) of five show the same pattern as the mother (Bl), despite coming from male fetuses (Am). DNA in Lanes 1 and 2 could not be amplified.
Discussion
Because of the rarity of fetal NRBCs in maternal peripheral blood, it has been a challenge to enrich fetal cells from maternal peripheral blood and then analyze them for genetic disorders.
In this study, we used CD71 antibody against the transferrin receptor, which has been widely employed. In addition to MACS with the anti-CD71 antibody, we further tried to differentiate fetal NRBCs from other cells by anti-GPA antibody and by single-cell pick-up with a micromanipulator.
However, this study was fraught with considerable amplification failure (48.1%). There appeared to be several explanations for amplification failure. First, some template degradation may have occurred during the course of the enrichment and staining procedures (Reading et al. 1995; Cheung et al. 1996). Second, microdissection of cells fixed on slides can induce mechanical DNA breakage and thus loss of amplification from one or two alleles (Cheung et al. 1996). In these situations, however, analysis of several cells could avoid misdiagnosis. Third, it is probably to some extent related to cell loss during the micromanipulation, because the final transfer of the picked cell to the PCR tube is not under visual control. Laser-assisted micromanipulation and automatic transfer directly into the PCR tube (Schutze and Clement-Sengewald 1994) might circumvent cell loss and mechanical DNA breakage. Finally, although we used PEP, it is impossible to amplify the target sequences every time because of significant locus-dependent variation in amplification efficiency (Sekizawa et al. 1996b).
Despite these problems, we could correctly assign fetal gender in 13/20 (65%) pregnant women. This result is comparable to those of previous studies (Sekizawa et al. 1996a,b; von Eggeling et al. 1997).
Because we used heterozygous alleles (amelogenin gene and BclI polymorphism) and whole-genome amplification, we expected to have allelic drop-out (ADO).
As many researchers (von Eggeling et al. 1997; Garvin et al. 1998; Hahn et al. 1998) have observed, ADO is more likely to occur after whole-genome amplification. Nevertheless, Hahn et al. (1998) and Garvin et al. (1998) have recommended examination of multiple single cells and multiplex PCR for simultaneous identification using microsatellite loci known to be informative for solving the problem of ADO. Accordingly, we performed PCR in 47 cells and performed PCR for the amelogenin gene and BclI polymorphism sequentially to avoid ADO. Nevertheless, only one male-bearing pregnancy was informative in BclI polymorphism, and because of the relatively high failure rate in amplification, we cannot report the exact number of cells showing ADO.
Interestingly, three cells in this case showed a female-specific band in amelogenin PCR despite a male-bearing pregnancy and showed the same band pattern as that of the mother in BclI polymorphism as well. Combining these results, we could conclude that these three cells were of maternal origin. This finding also gives evidence that NRBCs in maternal blood are of both fetal and maternal origin, and it is comparable to those of previous studies (Ganshirt et al. 1994; Slunga-Tallberg et al. 1995,1996; von Eggeling et al. 1997; Holzgreve et al. 1998; Troeger et al. 1999).
It was not possible within the scope of this study to obtain large-scale results from BclI polymorphism, and therefore, because of the lack of informative cases, we could not demonstrate the validity of prenatal diagnosis of hemophilia A in our approach. However, with a larger number of cases that are informative in BclI polymorphism, we could identify patients affected by hemophilia A using fetal NRBCs in maternal blood.
In summary, our data demonstrate that our experimental design is valid and that if we apply our approach to more cases that are informative in BclI polymorphism, it will be possible to use fetal NRBCs in maternal blood for prenatal diagnosis of hemophilia A.
Footnotes
Acknowledgements
This study was supported by a grant from the Korea Health 21 R&D Project, Ministry of Health and Welfare, Republic of Korea (01-PJ10-PG6–01GN13–0002).