
Abstract
The prodeath effects of p53 are typically mediated via its transcriptional upregulation of proapoptotic Bcl-2 family members, including PUMA, Noxa, and/or Bax. We previously reported that staurosporine (STS), a broad-spectrum kinase inhibitor and prototypical apoptosis-inducing agent, produced p53-dependent, Bax-dependent, neural precursor cell (NPC) apoptosis, but that this effect occurred independently of new gene transcription and PUMA expression. To further characterize the mechanism by which p53 regulates NPC death, we used primary cerebellar NPCs derived from wild-type, p53-deficient, and Bax-deficient neonatal mice and the mouse cerebellar neural stem cell line, C17.2. We found that STS rapidly increased p53 cytoplasmic immunoreactivity in neuritic-like processes in C17.2 cells, which preceded Bax activation and caspase-3 cleavage. Confocal microscopy analysis of STS-treated cells revealed partial colocalization of p53 with the mitochondrial marker pyruvate dehydrogenase as well as with conformationally altered “activated” Bax, suggesting an interaction between these proapoptotic molecules in triggering apoptotic death. Nucleophosmin (NPM), a CRM1-dependent nuclear chaperone, also exhibited partial colocalization with both activated Bax and p53 following STS treatment. These observations suggest that cytoplasmic p53 can trigger transcription-independent NPC apoptosis through its potential interaction with NPM and activated Bax.
P
The tumor suppressor protein p53 is a key regulator of cell death under multiple physiological and pathological conditions. DNA-damaging agents, hypoxia, oxidative stress, and excitotoxic stimuli all induce p53-dependent death in the nervous system (Morrison et al. 2003). p53 is highly expressed in neural precursor cells (NPCs) in the embryonic ventricular zone, and a subset of p53-deficient mice show neural tube defects and hindbrain exencephaly (Armstrong et al. 1995; Sah et al. 1995). Compared with postmitotic neurons, NPCs, which include multipotent neural stem cells and lineage-restricted neural progenitor cells (Svendsen and Smith 1999), express relatively high levels of p53 (Meletis et al. 2006).
p53 has been shown to have dual mechanisms for inducing cell death. Some death stimuli, such as oxidative stress and genotoxic injury, cause p53 nuclear accumulation and activation of downstream proapoptotic gene expression, e.g., PUMA, Noxa, and/or Bax, to induce cell death (Mattson et al. 1999; Uo et al. 2007). Studies have suggested that genotoxic injury can also produce a “fast-wave” of cytoplasmic p53 that interacts with Bcl-2 family members to initiate transcription-independent apoptosis (Manfredi et al. 2003). However, the mechanism by which cytoplasmic p53 induces cell death is still unclear. We previously showed, in primary cerebellar NPCs, that genotoxin-induced NPC death is p53 dependent and requires new protein synthesis and expression of PUMA, a p53 transcriptionally regulated proapoptotic BH3-only molecule. In contrast, staurosporine (STS)-induced NPC death does not require new protein synthesis or PUMA expression. Similar results were found in C17.2 cells, a neural stem cell line derived from the mouse cerebellum, i.e., genotoxic agents increased nuclear p53 immunoreactivity (IR), whereas STS caused rapid cytoplasmic p53 accumulation. In this study, we further investigated p53-dependent, transcription-independent NPC death by examining the time course and subcellular localization of p53 and activated Bax IR in NPCs after STS exposure. We found that STS promoted rapid p53 cytoplasmic accumulation independent of CRM1, a previously described mediator of p53 nuclear export. In the cytoplasm, p53 exhibited partial mitochondrial colocalization with activated Bax, which preceded downstream caspase-3 activation and apoptosis. Cytoplasmic p53 also exhibited colocalization with nucleophosmin (NPM), a nuclear chaperone protein previously implicated in STS-induced cell death. These studies provide new insights into the potential molecular mechanisms of p53-dependent cell death.
Materials and Methods
Chemicals
STS, cytosine arabinoside (AraC), bleomycin, and lactasystin were purchased from Sigma (St. Louis, MO). BOC-aspartyl(Ome)-fluoromethyl ketone (BAF) was purchased from MP Biomedicals (Aurora, OH). Leptomycin B (LMB) was purchased from LC Laboratories (Woburn, MA).
Mice
Generation of bax−/- mice (Knudson et al. 1995) has been described previously. p53+/- mice were purchased from Taconic (Germantown, NY). Endogenous and disrupted genes were detected by PCR analysis of tail DNA extracts as described previously (Akhtar et al. 2006). The day the pups were born was counted as day 0. Mice were cared for in accordance with the guidelines of the NIH Guide for the Care and Use of Laboratory Animals. All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
Cell Cultures
Cerebellar NPCs were harvested from the cerebellum of postnatal day 7 mice and expanded in fibroblast growth factor-2 (FGF-2) as previously described (Geng et al. 2007). Briefly, cells were placed in uncoated flasks (Corning, Inc.; Corning, NY) at 37C in a humidified 5% CO2/95% air atmosphere, and glial cells, postmitotic neurons, and other adherent cell populations were allowed to attach to the bottom of the flask. Twenty-four hr later, floating cells, containing an enriched population of NPCs, were transferred to poly-
The C17.2 cells were cultured in high-modified DMEM (Gibco; Grand Island, NY) containing 1% penicillin/streptomycin, 1%
Cell Viability Assays
As previously described (Nowoslawski et al. 2005; Shacka et al. 2006), cells were washed once with Locke's buffer, then incubated at 37C for 30 min in Locke's buffer containing 5 μM calcein-AM (Molecular Probes; Eugene, OR). Calcein-AM conversion was measured using a fluorescence plate reader (excitation 488 nm, emission 530 nm). Calcein-AM conversion was expressed relative to untreated controls.
Immunocytochemistry (ICC)
For immunocytochemical detection, cells were fixed in 4% paraformaldehyde for 30 min at 4C, washed three times with PBS, then incubated for 30 min in PBS blocking buffer [(PBS-BB) 1 × PBS containing 1% BSA, 0.2% powdered milk, and 0.3% Triton X-100]. Primary antibodies were diluted in PBS-BB (without Triton X-100) at the indicated dilutions and applied overnight at 4C. Primary antibodies used were anti-p53 [(1:5000) Ncl-p53-Cm5P rabbit polyclonal anti-serum; Novocastra Laboratories, Newcastle upon Tyne, UK], anti-activated Bax [(1:1000) BD Pharmigen; San Jose, CA], anti-cleaved caspase-3 [(1:1000) Cell Signaling; Danvers, MA], anti-pyruvate dehydrogenase [(anti-PDH) (1:1000) mouse monoclonal, Molecular Probes; or rabbit anti-PDH (1:1000) Cell Signaling], and anti-NPM [(1:2000) Zymed; Carlsbad, CA]. Following washes with PBS, cells were incubated with horseradish peroxidase (HRP)-conjugated donkey anti-rabbit or anti-mouse secondary antibodies (Jackson ImmunoResearch; West Grove, PA), diluted to 1:2000 in PBS-BB (without Triton X-100), for 1 hr at room temperature. Following washes with 1XPBS, IR was detected using tyramide signal amplification [(TSA) Perkin-Elmer Life Science Products; Boston, MA] according to the manufacturer's instructions. Cultures were counterstained with bisbenzimide (2 μg/ml, Hoechst 33258; Sigma) and examined with a Zeiss-Axiovert fluorescence microscope (Jena, Germany). To quantitate IR, the number of positive cells was counted as a fraction of total cells in four representative fields for each well. The specificity of the p53, Bax, and caspase-3 immunocytochemical detection was verified using cultures derived from p53-, Bax-, and caspase-3–deficient mice, respectively. In each case, no IR was observed in cells lacking the relevant protein. Additional immunocytochemical controls consisted of omission of primary and/or secondary antibodies and substitution of non-immune serum or irrelevant antibodies.
For dual labeling of p53 and activated Bax or p53 and NPM, after TSA detection of p53 using rabbit polyclonal antiserum, 0.3% H2O2 was added for 10 min to destroy any residual HRP activity, followed by PBS washes. Cells were incubated for 30 min in PBS-BB, followed by diluted activated Bax or NPM mouse monoclonal antibodies in PBS-BB without Triton overnight. The next day, goat anti-mouse Vector Impress (Vector Laboratories; Burlingame, CA) was applied for 1 hr, followed by TSA detection (Perkin-Elmer Life Science Products). The dilutional neglect method was utilized to assess colocalization of activated Bax IR and NPM IR (Shindler and Roth 1996). TSA detection as described above was first performed for activated Bax, followed by PBS-BB for 30 min and overnight incubation with diluted NPM antibody in PBS-BB without Triton. The following day, cells were washed in PBS and incubated for 1 hr in diluted fluorescein-conjugated anti-mouse secondary antibodies (Jackson ImmunoResearch). Appropriate controls were performed to ensure that the activated Bax antibody was sufficiently dilute to be undetectable by the fluorescein-conjugated secondary antibody used to detect NPM IR.
Confocal Microscopy
For dual-label confocal imaging, cells on chamber slides were immunocytochemically labeled as described above, stained with bisbenzimide, coverslipped, and imaged using a Leica Confocal TCS SP1 ultraviolet (UV) unit with a Coherent Laser Group Enterprise (Santa Clara, CA) UV laser for blue fluorochromes, argon laser for green fluorochromes, and helium/neon laser for far-red fluorochromes. Filter sets for fluorescein and Cy3 were used (Chroma Technology Corp.; Brattleboro, VT). Fluorochrome excitation and emission were controlled by using an accusto optical tunable filter and prism spectrophotometer. The 100x objective and Leica confocal software were used to acquire images from untreated and STS-treated cells.
Cell Fractionation
Preparation of nuclear and cytoplasmic fractions was performed using the NE-PER Pierce Biotechnology Kit (Pierce; Rockford, IL) according to the manufacturer's instructions. Briefly, cells were suspended in reagent I (Cer I) containing 1% protease inhibitor cocktail (Sigma) and 1% phosphatase inhibitor cocktail (Sigma) and then incubated on ice for 10 min. Eleven ml of reagent II (Cer II) was added to the mixture, vortexed, and then incubated on ice for 1 min. The sample was centrifuged at maximum speed (13,000 × g) for 10 min at 4C. The supernatant (cytoplasmic fraction) was transferred to a new tube and stored at −80C. The pellet was resuspended in 100 ml of ice-cold nuclear extraction reagent (NER I) and vortexed for 15 s every 10 min, for a total of 40 min. The sample was then centrifuged at maximum speed (13,000 × g) for 10 min at 4C. The supernatant (nuclear fraction) was transferred to a new tube and stored at −80C. Protein concentrations were determined via a Pierce BCA kit.
Western Blot
Preparation of whole-cell lysates was performed as follows. Briefly, cells were suspended in lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, and 10% glycerol) with 1% PMSF, 1% protease inhibitor cocktail (Sigma), and 1% phosphatase inhibitor cocktail (Sigma) and incubated on ice for 30 min. Samples were centrifuged for 10 min at 13,000 × g, and the supernatant was transferred to a fresh tube. The deubiquitinase inhibitor N-ethylmaleimide (Sigma) was added to cell lysates harvested for p53 ubiquitination detection. The lysates were resuspended with 5x sample buffer and boiled for 10 min. Proteins were then resolved in a 10–15% SDS-PAGE gel and transferred to a nitrocellulose membrane for Western blot detection. Primary antibodies used were anti-poly (ADP-ribose) polymerase-1 (PARP) (Cell Signaling, 9542), anti-p53 (Cell Signaling, 2524), anti-phospho p53 (Ser18) (Cell Signaling, 9284), anti-activated Bax (BD Pharmigen), and anti-NPM (Zymed). Following incubation with appropriate secondary antibodies [anti-mouse (Cell Signaling); anti-rabbit (BioRad)], signal was detected using an enhanced chemiluminescence Western blotting analysis system (Amersham; Buckinghamshire, UK) or super-signal chemiluminescence (Pierce).
Statistics
All data points represent mean ± SEM; n=6 wells for all experiments. All experiments were repeated at least three times unless stated otherwise. Representative data are shown. Genotype-specific effects of treatment were analyzed for significance via two-way ANOVA. Post hoc analysis was conducted using Bonferroni's test. A level of p<0.05 was considered significant.
Results
STS Promotes p53 Accumulation in Neuritic-like Processes in C17.2 Cells
We have previously shown that C17.2 cells, a mouse cerebellar neural stem cell line, share similar death pathways with primary mouse NPCs after STS and genotoxic injury. After STS treatment of C17.2 cells, there is a significant amount of p53 IR accumulated in cytoplasmic “blebs” (Geng et al. 2007). To further identify the relationship between p53 and cytoplasmic swellings, phase-contrast images were taken of untreated and STS-treated C17.2 cells. Untreated C17.2 cells had a flattened, polygonal shape, whereas STS-treated C17.2 cells underwent a phenotypic alteration and exhibited multiple thin, elongated processes, often containing large terminal swellings (Figure 1A). In untreated C17.2 cells, p53 IR, albeit present at only low levels, was predominantly found in the nucleus, with lesser amounts found in the cytoplasm. After STS treatment, the vast majority of p53 IR was non-nuclear and often localized to swellings at the ends of the neuritic-like processes (Figure 1A). To quantify cytoplasmic p53 levels, cytoplasmic and nuclear fractions were prepared from untreated and STS-treated (0.3 μM) cells. Western blot analysis for PDH was performed to confirm cytoplasmic fractions and for PARP to confirm nuclear fractions. C17.2 cells exposed to STS for 6 hr revealed a significant increase in cytoplasmic p53 levels, in comparison to untreated cells (Figure 1B).
STS-induced p53 Cytoplasmic Translocation Is CRM-1 Independent
Previous studies have shown that monoubiquitination of p53 may cause p53 translocation from the nucleus to the cytosol. To determine whether STS-induced p53 cytoplasmic accumulation resulted from a unique alteration in p53 ubiquitination, the pattern of p53 bands on Western blot, indicative of different levels of mono- and polyubiquitination, was measured after STS (0.3 μM), AraC (3 μM), or bleomycin (0.03 UN/ml) treatment, in the presence or absence of the proteasome inhibitor lactasystin (10 nM) for 6 hr. p53 protein levels were detected by Western blot analysis with a specific monoclonal antibody, and ubiquitination was assessed via the appearance of higher-molecular-weight isoforms. Without cotreatment with lactasystin, there was no increase of higher-molecular-weight p53-ubiquitinated isoforms after STS, AraC, or bleomycin treatment. Lactasystin treatment alone increased the levels of both p53 and ubiquitinated p53. Treatment with AraC or bleomycin, together with lactasystin, significantly increased p53 mono- and polyubiquitination levels (Figure 2A). Compared with lactasystin treatment alone, combined STS and lactasystin treatment had no additional effect on ubiquitinated p53 levels (Figure 2A). Our results provide no evidence for an STS-specific change in the p53 ubiquitination pattern, suggesting that selective p53 monoubiquitination is not the cause of cytoplasmic p53 accumulation following STS exposure.
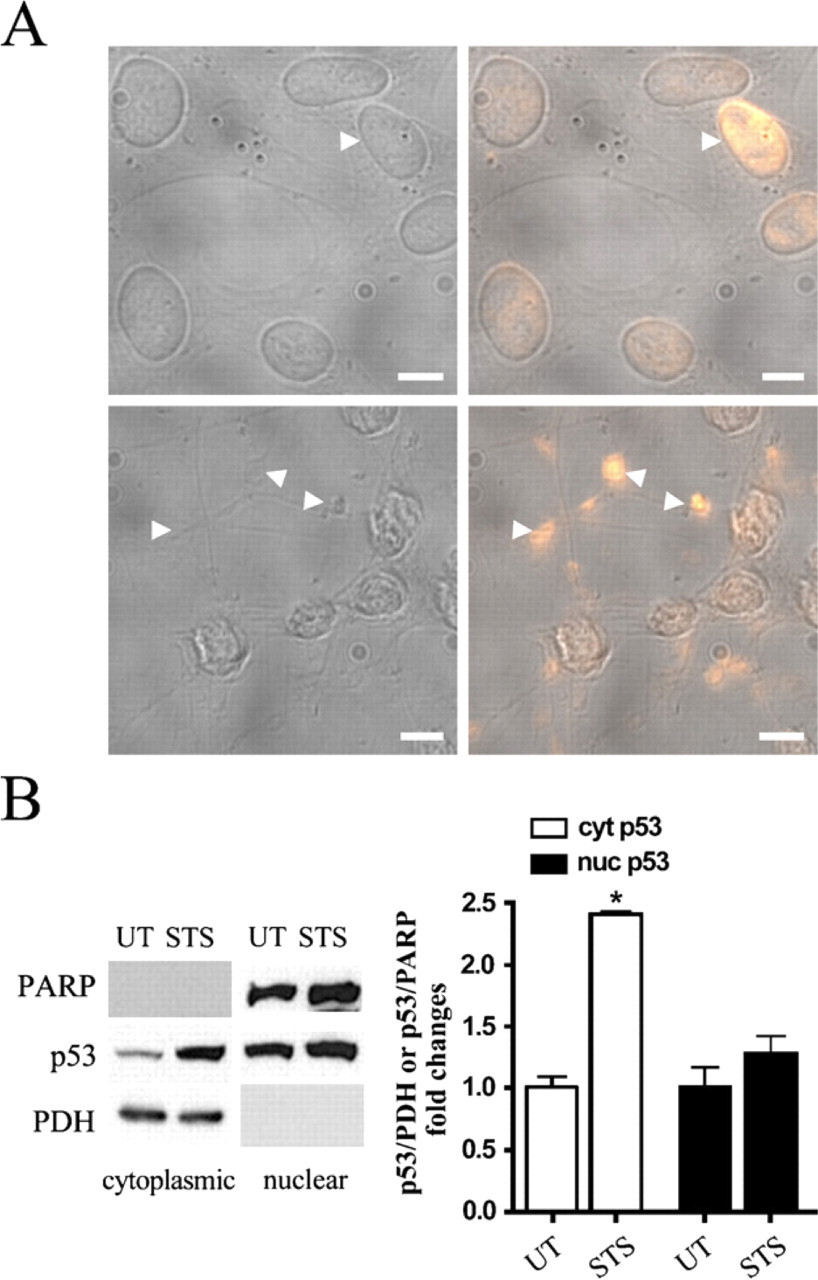
Staurosporine (STS) promotes p53 cytoplasmic accumulation in C17.2 cells. (
Because selective STS-induced ubiquitination of p53 does not obviously regulate p53 subcellular distribution, we hypothesized that STS might promote p53 cytoplasmic accumulation through a change in the nuclear export machinery. Previous studies have shown that CRM1 facilitates nuclear export of p53 and other proteins containing leucine-rich nuclear export signals (NESs) (Freedman and Levine 1998). LMB, an inhibitor of CRM1-dependent nuclear export, was added to C17.2 cells with or without STS treatment, and p53 was localized using ICC. Compared with untreated C17.2 cells, LMB (10 nM, 6 hr) significantly increased p53 nuclear IR (mean percentage of cells with nuclear p53 ± SEM: untreated, 8.9 ± 2.3% vs LMB-treated, 53.5 ± 8.5%; p<0.05). Pretreatment with 10 nM LMB for 6 hr followed by coadministration of 10 nM LMB and 0.3 μM STS for an additional 3 hr failed to inhibit STS-induced p53 nuclear-to-cytoplasmic trans-location (mean percentage of cells with nuclear p53 ± SEM: STS alone, 11.1 ± 5.1% vs STS plus LMB, 7.7 ± 1.14%; p>0.05) (Figure 2B). In summary, STS-induced p53 cytoplasmic accumulation was not inhibited by LMB; thus, it appears to be independent of CRM1-dependent nuclear export.
STS-induced p53 Cytoplasmic Accumulation Precedes Bax and Caspase-3 Activation
To test whether cytoplasmic p53 triggers the downstream apoptotic cascade, C17.2 cells were used to assess the temporal and spatial relationship between cytoplasmic p53 accumulation and apoptosis-associated intracellular events after STS treatment. C17.2 cells were treated with 0.3 μM STS for 0, 1, 2, 3, 4, or 6 hr; and p53, conformationally altered Bax, and cleaved caspase-3 were detected by Western blot analysis. Whole-cell lysates from STS-treated C17.2 cells revealed a significant increase in total p53 and activated Bax by 2 hr and cleaved caspase-3 by 3 hr, compared with untreated cells (Figure 3). These findings are consistent with our previous investigation of STS-induced NPC death, which indicated that caspase-3 activation following STS was dependent on p53 and Bax expression (Geng et al. 2007).
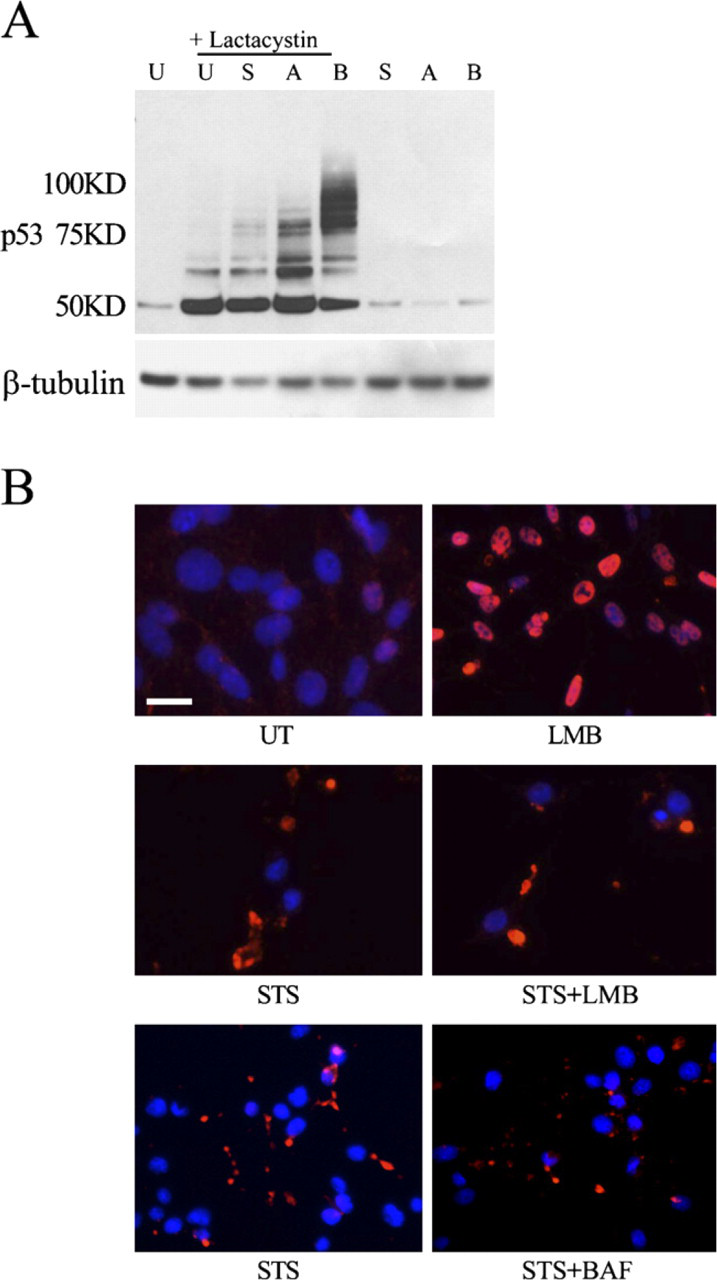
STS-induced p53 cytoplasmic accumulation is CRM-1 independent. (
Although cytoplasmic p53 is present before caspase-3 activity is detected in STS-treated cells, to rule out the possibility that activated caspases cleave nuclear lamin and induce nonspecific leakage of nuclear p53 into the cytoplasm, C17.2 cells were treated with 0.3 μM STS with or without 100 μM of the pan-caspase inhibitor BAF for 3 hr. Compared with STS treatment alone, BAF did not prevent p53 cytoplasmic accumulation (Figure 2B, bottom panel). Combined with our previous results indicating that cytoplasmic p53 preceded cleaved caspase-3 immunocytochemical detection, these results clearly show that STS-induced p53 cytoplasmic accumulation is not secondary to activation of caspases.
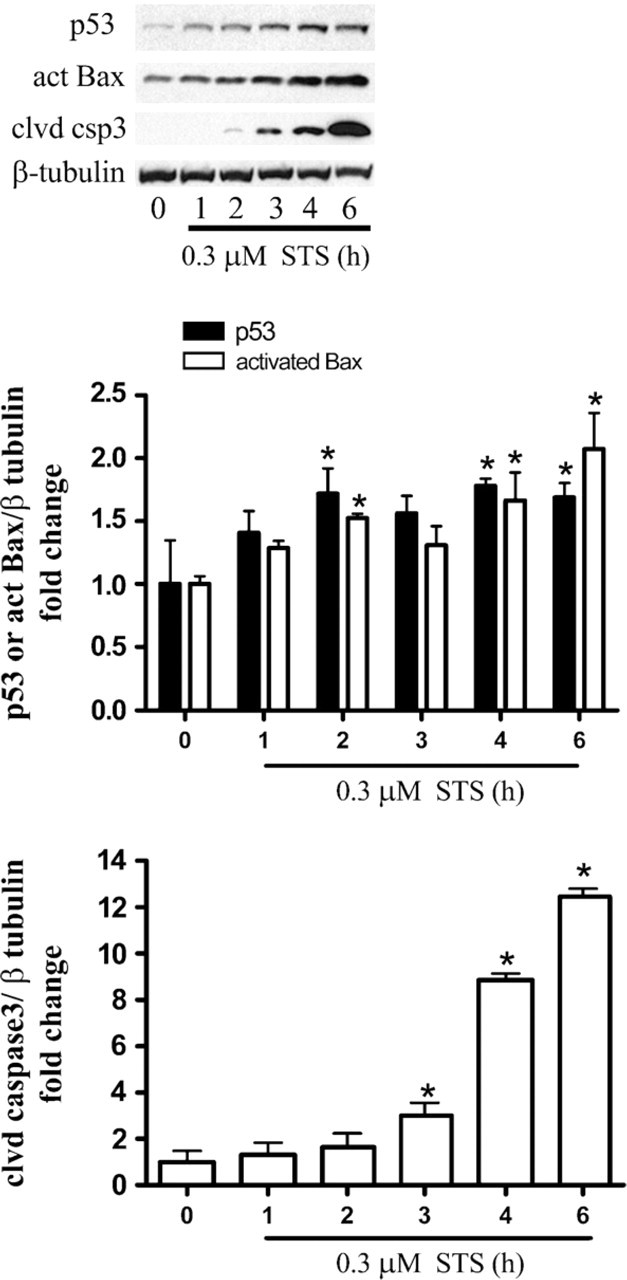
Staurosporine (STS) exposure results in a time-dependent increase in p53, activated Bax, and cleaved caspase-3. Biochemical detection revealed a significant increase in p53 and activated Bax as early as 2 hr after 0.3 μM STS exposure in C17.2 cells. Cleaved caspase-3 levels were significantly elevated at 3 hr and persisted for several hours. Western blots were digitized by UN-SCAN-IT software; p53, activated Bax, or cleaved caspase-3/β-tubulin pixel totals were determined and normalized to UT groups, and values were expressed as fold change. The data represent mean ± SEM, with n=4. ∗ p< 0.01 by two-way ANOVA/Bonferroni's post hoc test vs UT.
Cytoplasmic p53 and Activated Bax Show Colocalization With the Mitochondrial Marker PDH
The subcellular localization of p53 was examined by dual-label ICC and digital confocal microscopy after STS treatment for 6 hr. Mitochondria were selectively labeled using antibodies recognizing PDH, a mitochondrial matrix protein associated with the inner mitochondrial membrane (Odin et al. 2001). In contrast to untreated NPCs, which showed only low levels of p53 without obvious mitochondrial colocalization, STS treatment led to partial colocalization of p53 with PDH (Figure 4A). The majority of p53 IR, however, was unassociated with PDH IR and is likely to be localized to the cytosol.
We also performed dual-label ICC analysis for activated Bax and PDH to determine whether activated Bax localized to the mitochondria following STS treatment. Our results revealed that activated Bax IR was robust in STS-treated cells, compared with untreated cells, and showed near total colocalization with PDH (Figure 4B), which is consistent with the known association of confirmationally altered Bax with mitochondrial membranes.
STS Promotes the Interaction Between p53 and Activated Bax
Previous studies have shown that p53 may interact with Bcl-2 family members to induce cell death. We hypothesized that cytoplasmic p53, either through an interaction with conformationally altered Bax at the mitochondria or with unactivated Bax in the cytosol, may promote Bax-dependent NPC death. Cerebellar NPCs were prepared from p53-deficient and wild-type littermate mice at postnatal day 7, expanded in FGF-2 in vitro, and then exposed to STS. p53 deficiency significantly decreased STS-induced cerebellar NPC death (Figure 5A). Similarly, compared with cerebellar NPCs prepared from wild-type mice, Bax-deficient cerebellar NPCs exhibited significantly less STS-induced death (Figure 5B). Together, these data indicate that both p53 and Bax are critical regulators of NPC death induced by STS and confirm our previous report (Geng et al. 2007). To test whether p53 induced Bax activation, p53-deficient cerebellar NPCs, as well as wild-type NPCs, were treated with 0.3 μM STS for 3 hr, and ICC detection of activated Bax was performed. Quantification of the cells with positive activated Bax IR showed that compared with wild-type NPCs (24.9 ± 7.02%), p53 deficiency (2.2 ± 0.75%) significantly decreased Bax activation after STS exposure (p<0.05), indicating that p53 is an important regulator of Bax activation.
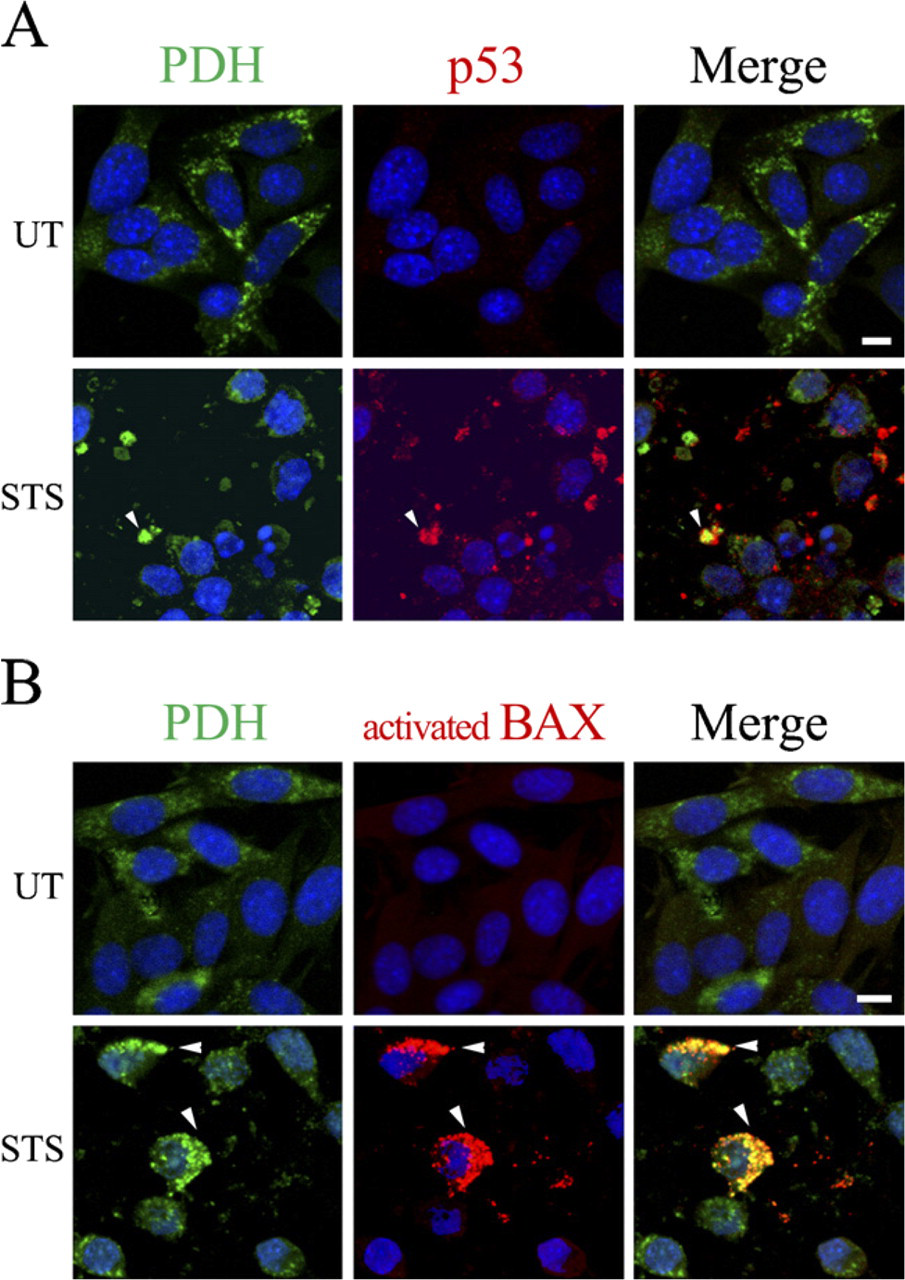
p53 and activated Bax colocalize with the mitochondrial marker PDH after staurosporine (STS) treatment. (
On the basis of the results of our ICC studies, showing that cytoplasmic p53 accumulation preceded activated Bax immunodetection and that p53 showed both cytosolic and mitochondrial localization, whereas activated Bax IR was predominantly mitochondria associated, we performed confocal microscopy to determine whether p53 and activated Bax colocalized. Confocal microscopy confirmed a partial overlap of p53 with activated Bax IR after STS treatment (0.3 μM, 6 hr) (Figure 6), suggesting that the interaction between p53 and activated Bax may regulate NPC death upon STS exposure. Although we were unable to perform triple labeling to determine whether the p53 and activated Bax IR colocalization occurred exclusively in mitochondria, the pattern of colabeling suggests that this was the case.
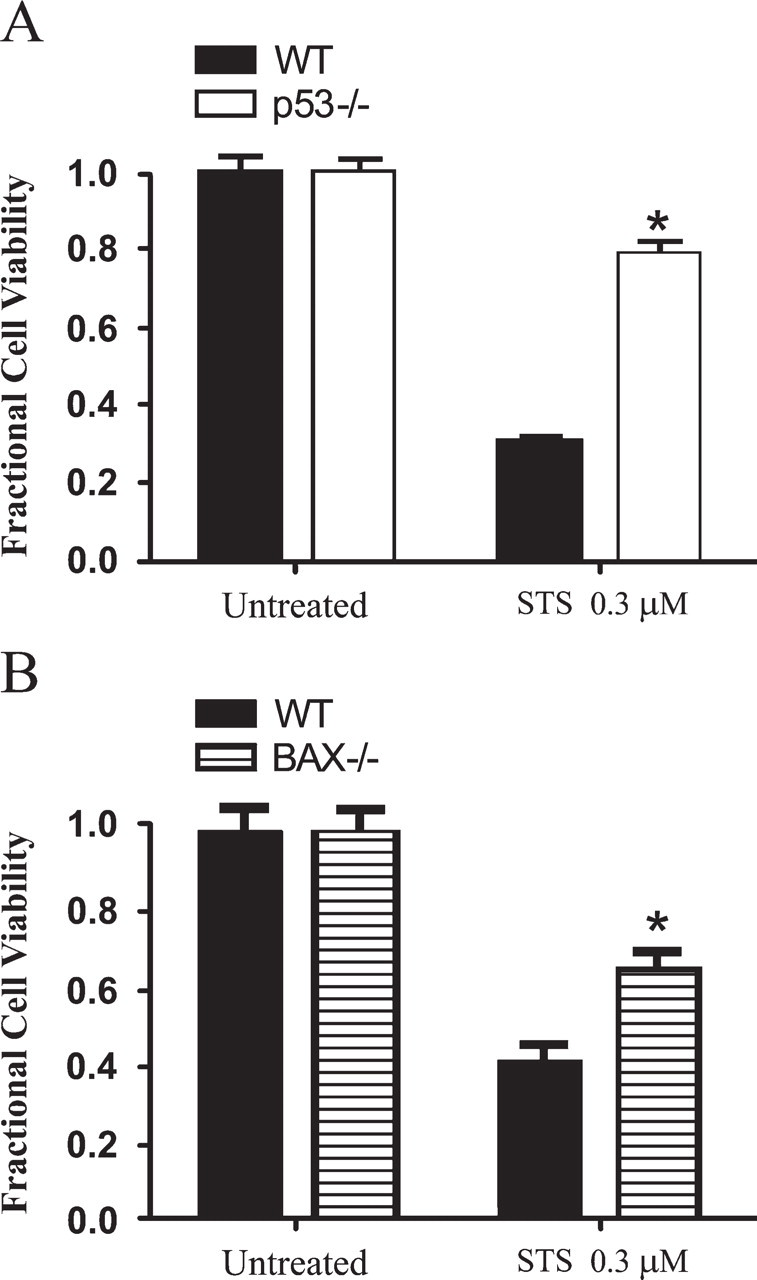
Staurosporine (STS)-induced neural precursor cell (NPC) death is p53 and Bax dependent. (
STS Promotes the Interaction of NPM With p53 and Activated Bax
NPM/B23 is a nucleolar phosphoprotein that is critical for nucleosome assembly and maintenance of chromatin structure (Eirin-Lopez et al. 2006; Frehlick et al. 2007). Like p53, nucleocytoplasmic shuttling of NPM has been shown to be mediated by CRM1 (Wang et al. 2005). Surface plasmon resonance studies have demonstrated that the C-terminal domain of NPM interacts with two regions of p53 (Lambert and Buckle 2006) and, in addition, that NPM serves as a novel Bax-binding protein during the apoptotic cascade. Here, we tested the hypothesis that STS promotes the interaction of NPM with p53 and/or activated Bax in NPCs. We performed confocal microscopy studies to detect the possible colocalization of NPM with activated Bax, as well as NPM with p53 after STS treatment. C17.2 cells were treated with 0.3 μM STS for 6 hr, and NPM IR and p53 IR were detected using ICC and confocal laser scanning microscopy. Compared with untreated control cells, there was increased cytoplasmic colocalization of NPM and p53 as well as NPM IR and activated Bax IR after STS exposure (Figures 7A and 7B). Overall, our results suggest that cytoplasmic NPM, p53, and activated Bax may interact with each other to promote cell death after STS exposure.
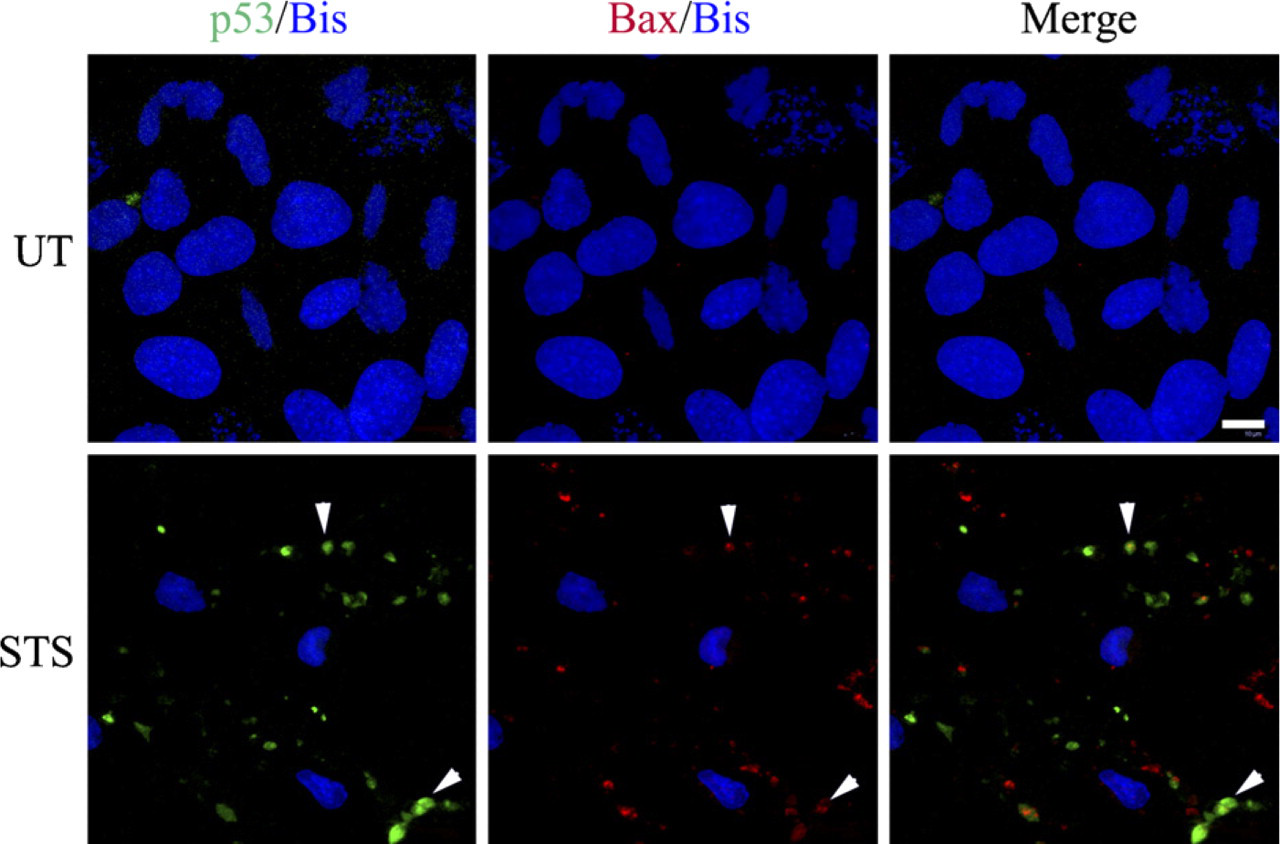
Staurosporine (STS) promotes p53 and activated Bax colocalization. Confocal images of untreated (UT) and STS-treated (0.3 μM, 6 hr) cells were taken to assess the relationship between activated Bax (cy3) and p53 (fitc) IR. Control cells exhibited only very low levels of either p53 or activated Bax, which were difficult to detect at baseline. STS treatment resulted in significantly increased p53 and activated Bax IR, which exhibited partial colocalization (arrowheads). Additional p53 IR was observed that did not colocalize with activated Bax and presumably localized to the cytosol. Bar = 10 μm.
Discussion
In this study, we explored the molecular regulation of p53-dependent, Bax-dependent, transcription-independent NPC apoptosis. We showed that STS promoted cytoplasmic p53 accumulation, which preceded Bax activation and caspase-3 cleavage. Confocal studies showed an increase in p53 and activated Bax colocalization following STS treatment. In addition, we found that STS caused p53 cytoplasmic accumulation through a CRM1-independent mechanism, which could not be inhibited by broad-spectrum caspase inhibition. NPM, a nuclear chaperone, has been shown by other investigators to be involved in the regulation of apoptosis, possibly through its interaction with conformationally altered Bax. Confocal microscopy studies also showed that STS promoted a partial colocalization between activated Bax and p53, activated Bax and NPM, as well as NPM and p53, suggesting that cytoplasmic p53, NPM, and activated Bax may interact with each other to induce apoptosis.
p53 can induce apoptosis through multiple mechanisms. Nuclear p53 can bind to DNA and activate proapoptotic gene expression; alternatively, cytoplasmic p53 can trigger transcription-independent apoptosis by directly interacting with Bcl-2 family members (Chipuk et al. 2005). Several models have been proposed to explain how cytoplasmic p53 interacts with Bcl-2 family members to induce cell death. One model suggests that p53 may translocate to mitochondria, where it binds to prosurvival Bcl-2 or Bcl-X
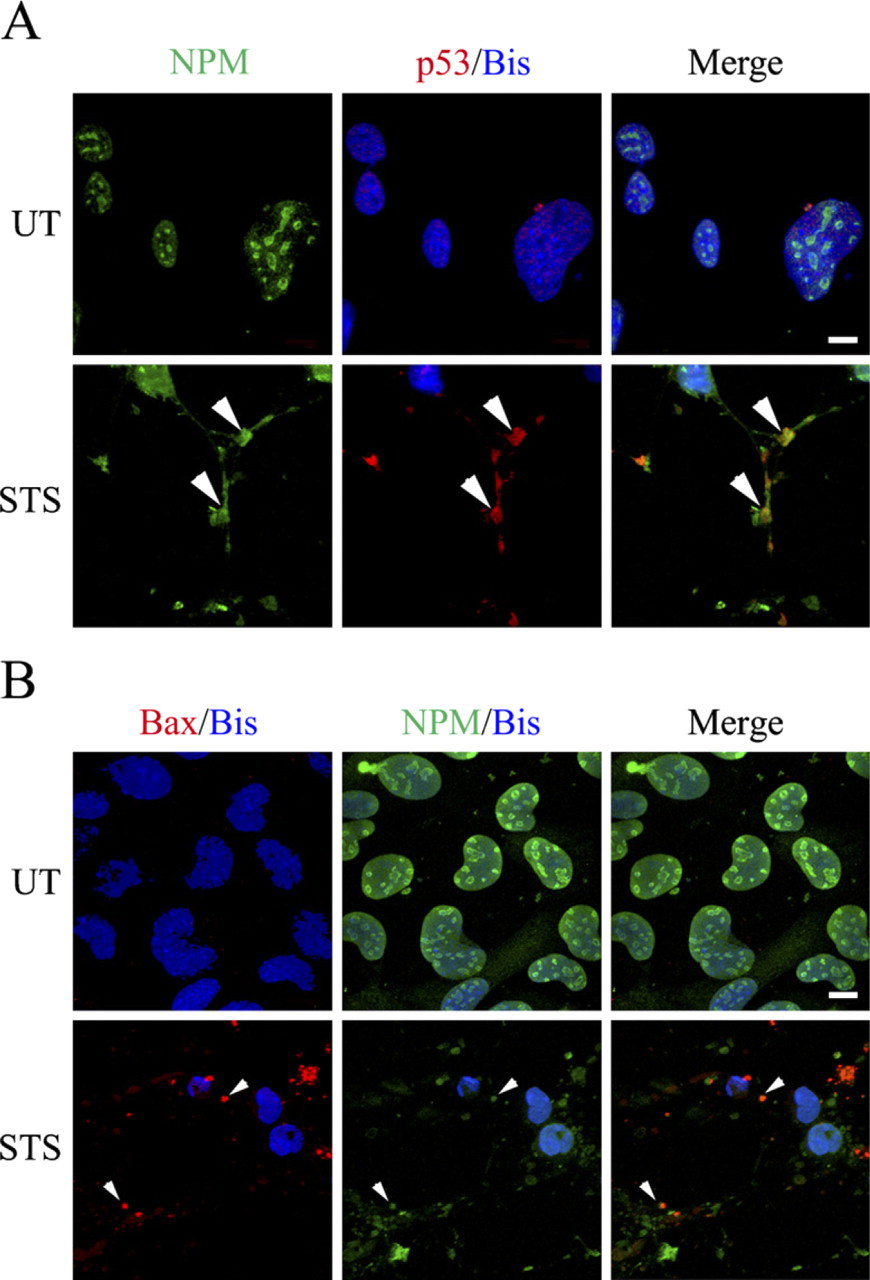
Staurosporine (STS) promotes nucleophosmin (NPM) colocalization with p53 and activated Bax. C17.2 cells were either left untreated (UT) or treated with 0.3 μM STS for 6 hr. (
How p53 targets to different subcellular compartments after specific death stimuli is largely unknown. It has been proposed that posttranslational modification of p53 may regulate its subcellular localization. Phosphorylation and acetylation of p53 sequester p53 in the nucleus (Appella and Anderson 2001), whereas monoubiquitination of p53 leads to its nuclear export (Li et al. 2003) or mitochondrial translocation. In our study, compared with the genotoxic agents AraC and bleomycin, STS did not specifically alter the ubiquitination pattern of p53 while at the same time producing p53 cytoplasmic accumulation. Previous studies have shown that caspases can cleave p53 into products that lack a nuclear localization signal, resulting in p53 becoming sequestered in the cytosol (Sayan et al. 2006). However, our results showed that p53 cytoplasmic translocation preceded caspase-3 activation and was not affected by pancaspase inhibition. Thus, cytoplasmic p53 in NPCs is not a direct consequence of the caspase activation induced by STS.
Movement of proteins between the nucleus and the cytoplasm may require interaction with nuclear pore complex components (Talcott and Moore 1999). Some proteins that shuttle between the nucleus and the cytosol independent of the nuclear pore complex may be controlled purely by their molecular size. Other nuclear proteins, especially those with molecular mass larger than 50 kDa, require interaction with nuclear pore components and/or intermediate carriers to escape the nucleus. The nuclear export receptor CRM1 regulates nuclear export of specific proteins by binding to NESs, and p53 is one such protein. Other studies and our experiments reported here show that a CRM1-specific inhibitor, LMB, induces nuclear p53 accumulation when added to cells under baseline conditions. However, LMB failed to inhibit STS-induced p53 cytosolic accumulation, indicating that STS-induced p53 nuclear-to-cytoplasmic accumulation is CRM1 indepdendent.
NPM is a member of the nuclear phosphoprotein family, which has been shown to translocate from nucleus to cytosol through CRM1-dependent nuclear export machinery. NPM is capable in vitro of physically interacting with a synthetic Bax peptide that mimics conformationally altered activated Bax, and this interaction may regulate apoptosis. NPM deficiency has been shown to attenuate STS-induced cell death in a human neuroblastoma cell line (Kerr et al. 2007), suggesting an important role for p53, Bax, and NPM interaction in response to this prototypical apoptotic stimulus. Our results showed that STS promoted colocalization of p53 and activated Bax, NPM and activated Bax, and p53 and NPM. Whether these three molecules form a tri-molecular complex or sequentially interact with each other to induce apoptosis is unclear and requires further investigation.
Defective p53-mediated apoptosis contributes to human tumor formation, and the p53 gene is the most commonly mutated gene in human cancers. Mutations in p53 also render cancer cells resistant to chemotherapy and radiation therapy. Therefore, restoration of p53-dependent cell death is an important therapeutic goal in clinical oncology. To date, attempts at restoring p53-dependent, transcription-dependent apoptosis have focused on small molecules such as CP-31398 and PRIMA-1 that structurally rescue mutated p53 proteins and maintain its transcriptionally active conformation (Foster et al. 1999). However, the DNA binding domain of p53 is commonly mutated in many tumors, and its transcriptional function is largely compromised. In contrast to its transcription-dependent function, the p53 transcription-independent apoptotic effect does not require the p53 DNA binding domain and could take advantage of preexisting proapoptotic molecules to induce tumor cell death (Chipuk and Green 2004). Thus, the therapeutic potential of cytoplasmic p53 as a tumoricidal mechanism has been proposed (Fuster et al. 2007). For example, in a transgenic mouse model of lymphoma, mitochondria-targeted p53 significantly induces tumor cell apoptosis in vivo (Talos et al. 2005). Nutlin-3a, an Mdm2 inhibitor, has been reported to promote p53-mediated apoptosis in chronic lymphocytic leukemia, at least in part via a transcription-independent mechanism (Kojima et al. 2006). Thus, STS-induced, p53 transcription-independent death may have therapeutic potential in non-neuronal tumors as well as in central nervous system tumors, e.g., human medulloblastoma, which have relatively high levels of p53 expression (Adesina et al. 1994, 2000). A detailed understanding of the mechanism by which STS causes apoptosis will be necessary to identify possible molecular targets for therapeutic development.
Footnotes
Acknowledgements
This work was supported by National Institutes of Health (NIH) Grants NS-35107, NS-41962, and CA-134773.
We thank Dr. Evan Snyder (Burnham Institute) for generously providing the C17.2 cell line, Cecelia B. Latham, the UAB Neuroscience Core Facilities (NIH NS-47466 and NS-57098), and Albert Tousson, Shawn Williams, and UAB Imaging Facility for technical assistance.