
Abstract
Thrombospondin-2 (TSP2) is an inhibitor of angiogenesis with pro-apoptotic and anti-proliferative effects on endothelial cells. Mice deficient in this matricellular protein display improved recovery from ischemia and accelerated wound healing associated with alterations in angiogenesis and extracellular matrix remodeling. In this study, we probed the function of TSP2 by performing a detailed analysis of dermal wounds and wound-derived fibroblasts. Specifically, we analyzed incisional wounds by tensiometry and found no differences in strength recovery between wild-type and TSP2-null mice. In addition, analysis of full-thickness excisional wounds by terminal deoxynucleotidyl transferase–mediated 2′-deoxyuridine 5′-triphosphate nick-end labeling stain and MIB-5 immunohistochemistry revealed similar numbers of apoptotic and proliferating cells, respectively. In contrast, the levels of matrix metalloproteinase (MMP)-2, MMP-9, tissue inhibitors of metalloproteinase (TIMP)-1, TIMP-2, and soluble vascular endothelial growth factor were increased in wounds of TSP2-null mice. Evaluation of the ability of TSP2-null wound fibroblasts to contract collagen gels revealed that it was compromised, even though TSP2-null wounds displayed normal myofibroblast content. Therefore, we conclude that the lack of TSP2 leads to aberrant extracellular matrix remodeling, increased neovascularization, and reduced contraction due in part to elevated levels of MMP-2 and MMP-9. These observations provide in vivo supporting evidence for a newly proposed function of TSP2 as a modulator of extracellular matrix remodeling.
T
Cutaneous wound healing is characterized by coordinated inflammation, reepithelization, tissue remodeling, and wound contraction at the injury site (Singer and Clark 1999). Attributed to forces generated by fibroblasts on granulation tissue, wound contraction is believed to lead to wound closure and scar formation (Bullard et al. 1999a, b; Bradshaw et al. 2002). A complex network of soluble biological mediators orchestrates wound healing including cytokines and growth factors (Singer and Clark 1999). These mediators are tightly regulated themselves by, among others, the matrix metalloproteinases (MMPs). The MMP protein family also influences remodeling of the provisional matrix and epithelial cell migration (Gill and Parks 2008). Knockout animal models have confirmed the importance of MMPs in wound healing, inasmuch as stromelysin-1 (MMP-3)-null mice display retarded wound healing related to decreased wound contraction (Bullard et al. 1999a, b). Regulation of MMPs can occur through zymogen activation, compartmentalization, and inhibition by the endogenous tissue inhibitors of metalloproteinases (TIMPs) (Ra and Parks 2007). Moreover, proper spatial distribution and regulation of MMPs are essential to wound healing, inasmuch as addition of the MMP inhibitor GM6001 delayed reepithelization (Mirastschijski et al. 2004; Gill and Parks 2008).
The spatiotemporal expression of TSP2 in wound healing is predominantly associated with the late proliferation/remodeling phase, and its maximum peak coincides with the onset of vascular regression, after which the levels of TSP2 drop nearly to background (Kyriakides et al. 1999b; Agah et al. 2002). The ability of TSP2 to control blood vessel formation in wounds has been demonstrated in studies exhibiting improved healing in wild-type (WT) mice by blockade of endogenous TSP2 synthesis or reversed vascular density of skin wounds, sponge granulomas, and foreign body capsules with exogenous expression of TSP2 in TSP2-null mice (Kyriakides et al. 1999a, b, 2001b). The exact mechanism by which TSP2 limits angiogenesis in vivo is not understood, but possible mechanisms include alterations in endothelial cell (EC) physiology by direct interaction with TSP2, through paracrine alterations exerted via altered cell–matrix interactions, and by changes in the extracellular milieu due to altered MMP profiles.
Evidence for alteration of EC physiology due to direct TSP2–receptor interactions includes previous in vitro studies demonstrating that TSP1 and TSP2 can induce either EC apoptosis directly (Guo et al. 1997; Jimenez et al. 2000; Nor et al. 2000) or cell cycle arrest in an apoptosis-independent fashion (Armstrong et al. 2002; Lopes et al. 2003; Oganesian et al. 2008). Alternatively, three-dimensional in vitro models of angiogenesis demonstrated blockade of EC tube formation by exogenous TSP2, but not by TSP1 (Krady et al. 2008). Supported by the observation that TSP2-null mice have altered ECM structure (Kyriakides et al. 1998), these observations have led to nascent investigations exploring paracrine ECM–cell interactions directed by TSP2. Although the mechanisms underlying altered ECM ultrastructure formation have not been elucidated, it is clear that ECM from TSP2-null mice alters EC behavior (Krady et al. 2008). Specifically, TSP2-null ECM is more permissive to EC migration and alters EC morphology during attachment. Collectively, these studies suggest that TSP2 might have an indirect, non–receptor-mediated role in modulating angiogenesis.
The role of TSP2 in recycling MMPs has emerged as a prominent mechanism for its anti-angiogenic function. TSP2-null fibroblasts have an intrinsic adhesive defect due to reduced clearance of MMP-2 from their extracellular environment (Yang et al. 2000). Specifically, TSP2 can bind MMP-2 and direct the complex to the scavenger receptor LRP-1, which is involved in the uptake and catabolism of TSPs (Godyna et al. 1995; Mikhailenko et al. 1995; Chen et al. 1996). The ability of TSP1 and TSP2 to interact directly with various MMPs in vitro has been reported (Bein and Simons 2000; Yang et al. 2000). In addition, the deficiency in TSP2 is associated with increased extracellular levels of MMP-2 in a variety of in vivo models (Kyriakides et al. 2001b; Kokenyesi et al. 2004; Schroen et al. 2004). More importantly, we recently showed that the increase in MMP-2 leads to a decrease in the levels of tissue transglutaminase, resulting in an alteration in the maturation of the wound matrix (Agah et al. 2005). Because of the established pro-angiogenic activity of MMPs, it is likely that increased angiogenesis in TSP2-null wounds is due, at least in part, to increased levels of MMP-2 and MMP-9. The latter has been shown to be effective in releasing matrix-bound vascular endothelial growth factor (VEGF), a pro-angiogenic growth factor, in a tumor model (Belotti et al. 2003). In separate studies, MMP-9, but not MMP-2, was shown to release matrix-bound VEGF (Bergers et al. 2000; Lee et al. 2005).
In the present study, we undertook a detailed analysis of the wound-healing response in TSP2-null mice, with a focus on the molecules and processes that might be influenced by TSP2. Specifically, increased MMP-2 and MMP-9, as well as soluble VEGF, were observed in TSP2-null wounds, which may account for observed increases in angiogenesis and altered ECM. Further, analysis of collagen gel contraction by wound fibroblasts demonstrated that the TSP2-null fibroblasts were inherently less contractile than WT cells, which may explain the improved cosmetic healing of wounds in TSP2-null mice. Collectively, our results demonstrate that TSP2-null wounds recover adequate tensile strength and implicate TSP2 in the regulation of the extracellular levels of MMP-2 and MMP-9 in skin wounds. Both MMP-2 and MMP-9 participate in the healing of skin wounds and are believed to be important mediators of this process (Parks 1999; Page-McCaw et al. 2007); thus, additional effects, including altered matrix remodeling in TSP2-null mice, could contribute to improved healing.
Materials and Methods
Animal Model
The generation of TSP2-null mice has been described (Kyriakides et al. 1998). In this study, unless stated otherwise, 3-month sex-matched TSP2-null and WT mice, both on a 129SvJ background, were used. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Washington and Yale University.
Wounds
Full-thickness excisional wounds were made in the dorsal region of mice anesthetized with Avertin, as described previously (Kyriakides et al. 1999b). Each mouse received two 6-mm wounds with the aid of a biopsy punch (Acuderm; Fort Lauderdale, FL), producing a total of 10 wounds per time point. All wounds were excised with a 3-mm rim of unwounded tissue. For the generation of wound extracts, a 3-mm biopsy punch was used to collect wound tissue from the center of each wound. For tensile strength analysis, two full-thickness longitudinal incisions, each 1.0 cm in length and separated by 1.5 cm, were made on the dorsal skin of each mouse with the aid of a standard #10 surgical blade. The first wound was administered on day 0 and the second on day 7. A total of six wounds per time point per genotype were made. All wounds were collected on day 14 and analyzed with the aid of an Instron 5542 tensiometer (Instron; Canton, MA), as described previously (Wu et al. 2003).
Tissue Processing and Immunohistochemistry
Excised wounds were fixed in 10% zinc-buffered formalin (Z-fix; Anatech, Battle Creek, MI) and embedded in paraffin. Sections 5-μm thick were generated and stained with various antibodies. Cells undergoing apoptosis/necrosis were detected using the terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate nick-end labeling (TUNEL) stain according to the supplier's instructions (Apoptag detection kit; Chemicon, Purchase, NY). Proliferating cells were detected using the MIB-5 antibody (DAKO; Carpenteria, CA) at 1:500 dilution, as described previously (Birner et al. 2001). This antibody recognizes the proliferation-specific antigen Ki-67 and has been shown to detect proliferating cells in paraffin-embedded murine tissues (Hollander et al. 2003) with the same efficiency as anti-BrdU antibodies (Birner et al. 2001). MMP-2 and MMP-9 were detected using antibodies (Chemicon; Temecula, CA) according to the supplier's instructions. Myofibroblasts were detected with pre-diluted anti–smooth-muscle actin antibody (Ab15267; Abcam, Cambridge, MA). Immune reactions, based on peroxidase activity, were visualized with the Vector ABC Elite kit (Vector Laboratories; Burlingame, CA). A Nikon Eclipse 800 microscope (Tokyo, Japan) equipped with fluorescence optics was used for all examinations.
Histomorphometry
Digital images from stained sections were collected and analyzed with the aid of imaging software (Metamorph; Universal Imaging Co., Downingtown, PA), as described previously (Kyriakides et al. 2001b). Briefly, the intensity and distribution of peroxidase activity (brown color) were quantified. Determination of MMP-2 and MMP-9 levels relied on the establishment of a threshold value, above which the detection of brown color was deemed to be reliable. To determine the appropriate threshold for MMP-2, sections were stained using an anti-human MMP-2 antibody that does not recognize mouse MMP-2 (ab52757; Abcam). MMP-9 threshold was determined by staining MMP-9-null wounds with the anti-MMP-9 antibody. MMP-9-null mice were a kind gift from Dr. Robert Senior (Washington University, St. Louis, MO). Values are given as arbitrary units and can only be used to compare results obtained with the same antibody. For the quantification of cells undergoing apoptosis/necrosis or proliferation, images were collected and analyzed independently by two investigators in a blind fashion. For all analyses, a total of 30 images per time point per genotype were analyzed.
Analysis of Wound Extracts
Protein extracts from day 7, day 10, and day 14 wounds were prepared in extraction buffer [PBS containing 2 mM/liter PMSF and 1 mg/ml each of aprotinin, leupeptin, and pepstatin (Sigma Chemical Co.; St. Louis, MO)] as described previously (Agah et al. 2002). The protein content of each sample was determined by the Bradford assay, according to the supplier's instructions (Bio-Rad; Hercules, CA). For zymography, equal amounts of protein were subjected to SDS-PAGE under non-reducing conditions in 7.5% acrylamide gels containing 0.1% gelatin. Zones of lysis were detected as described previously (Yang et al. 2000). A total of three samples per genotype were analyzed, and the experiments were repeated in triplicate. Western blot analysis of the same samples with anti-MMP-2 and anti-MMP-9 was performed as described previously (Yang et al. 2000). TIMP-1 antibody (1:200 dilution, SC5538; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and TIMP-2 antibody (1:1000 dilution, ab1828; Abcam) were employed for Western blot analysis and detected by an enhanced chemiluminescence Western blotting kit (Cat# RPN2108; Amersham Life Science, Piscataway, NJ). Soluble VEGF was prepared by gentle extraction of wound tissues in PBS containing 2 mM/liter PMSF and 1 mg/ml each of aprotinin, leupeptin, and pepstatin at 4C for 30 min. Western blot analysis was performed with anti-VEGF antibody at 0.2 μg/ml (Abcam; ab9953). Mouse VEGF-specific ELISA was performed with a Quantikine kit according to the supplier's instructions (R and D Systems; Minneapolis, MN).
Isolation of Wound Fibroblasts and Gel Contraction
Wound tissue (day 10) was obtained as described above, and fibroblasts were isolated as described previously (Kyriakides et al. 1998). Briefly, skin, including the wounded area collected with a 6-mm biopsy punch and wound tissue, was excised with the aid of a 3-mm biopsy punch. Three wounds per genotype were combined to generate wound fibroblasts, and specimens were cut into 0.5-mm2 fragments and allowed to adhere to the surface of 35-mm plates for 5 min. Cells were fed with DMEM supplemented with 10% fetal calf serum (FCS), and the medium was changed periodically until the explant-derived cells had become confluent. After two subcultures, the cell population appeared, morphologically, to be composed entirely of fibroblasts. No differences in the growth rates of TSP2-null and WT wound fibroblasts were observed. Wound fibroblasts (1 × 105 cells/ml) were mixed in a 1-mg/ml collagen (PureCol; Inamed Biomaterials, Fremont, CA) preparation (kept at 4C) and plated in 24-well dishes precoated with 1% agarose, and incubated at 37C/5% CO2. Three hr later, the solidified gels were fed with DMEM with or without 2% FCS. Gels were allowed to remain attached (stressed), or were relaxed by using a razor blade to separate the gel from the side of the plate. The diameter of relaxed gels was measured 48 hr later and converted to percent change from original area. Triplicate wells were used for each contraction assay, and the experiment was performed three times. Stressed gels were fixed in the wells with 4% paraformaldehyde, treated with 1% BSA/1% Triton X-100, stained with rhodamine-phalloidin (Molecular Probes; Carlsbad, CA) and visualized with the aid of a Nikon Eclipse microscope equipped with fluorescent optics. The experiment was performed twice.
Statistical Analysis
All comparisons between data sets were analyzed by the Student's
Results
Recovery of Normal Tensile Strength in TSP2-null Wounds
The reduced tensile strength of uninjured skin in TSP2-null mice (Kyriakides et al. 1998) suggested that the mechanical integrity of TSP2-null healing wounds might be compromised despite their improved appearance. To examine this possibility, we determined the tensile strength of day 7 and day 14 TSP2-null and WT incisional wounds. Wounds of both genotypes exhibited indistinguishable recovery of their tensile strength (Figure 1). Overall, the recovery between day 7 and day 14 was over 3-fold for each genotype. This finding suggests that TSP2-null wounds are not compromised with respect to the initial rate of accumulation and the quality of an extracellular matrix. Thus, despite the baseline reduced tensile strength of uninjured TSP2-null skin, wounds in these mice managed to assemble granulation tissue that provided normal tensile strength.
Normal Cellular Apoptosis and Proliferation in TSP2-null Wounds
Apoptotic and necrotic cells in excisional wounds of WT and TSP2-null mice were detected with TUNEL stain (Figure 2). The number of TUNEL-positive cells per high-power field decreased as the wounds matured, and no differences between TSP2-null and WT wounds were observed (Figure 2C). Similar to the findings for TUNEL, no difference in the number of proliferating cells per high-power field was observed between TSP2-null and WT wounds at any time point examined (Figure 2D). We were surprised by the lack of reduced cell death or increased proliferation in TSP2-null wounds, especially because TSP2-null wounds have been shown to have a higher cellular content than WT wounds (Kyriakides et al. 1999b). This apparent discrepancy might be explained by an increase in the recruitment, migration, or enhanced survival of repair cells in these wounds.
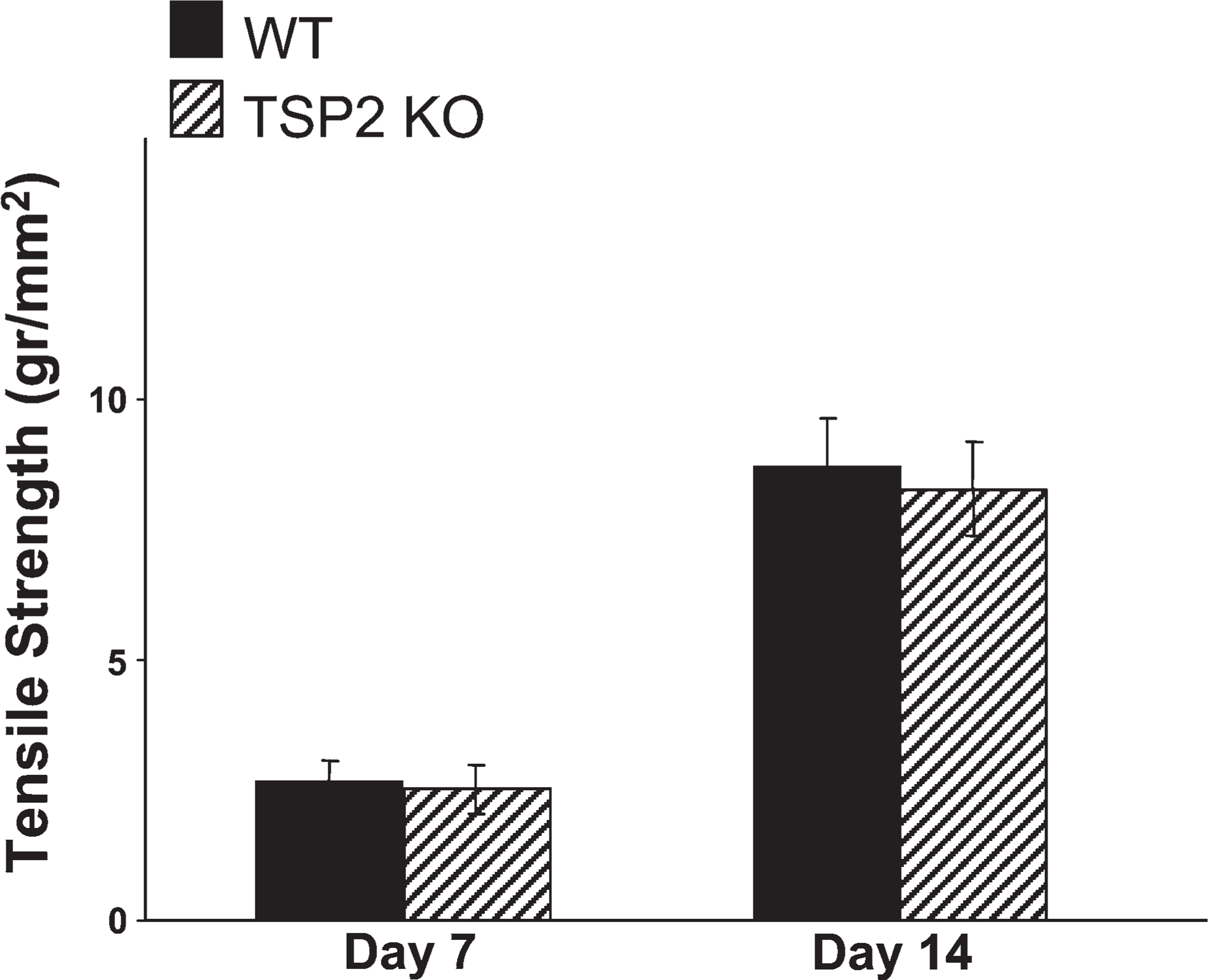
Recovery of normal tensile strength in thrombospondin-2 (TSP2)-null wounds. Samples of wild-type (WT) (black bars) and TSP2-null (hatched bars) wounds at 7 and 14 days of healing from mice 5 months of age were assessed for tensile strength with an Instron tensiometer. Error bars represent SEM (
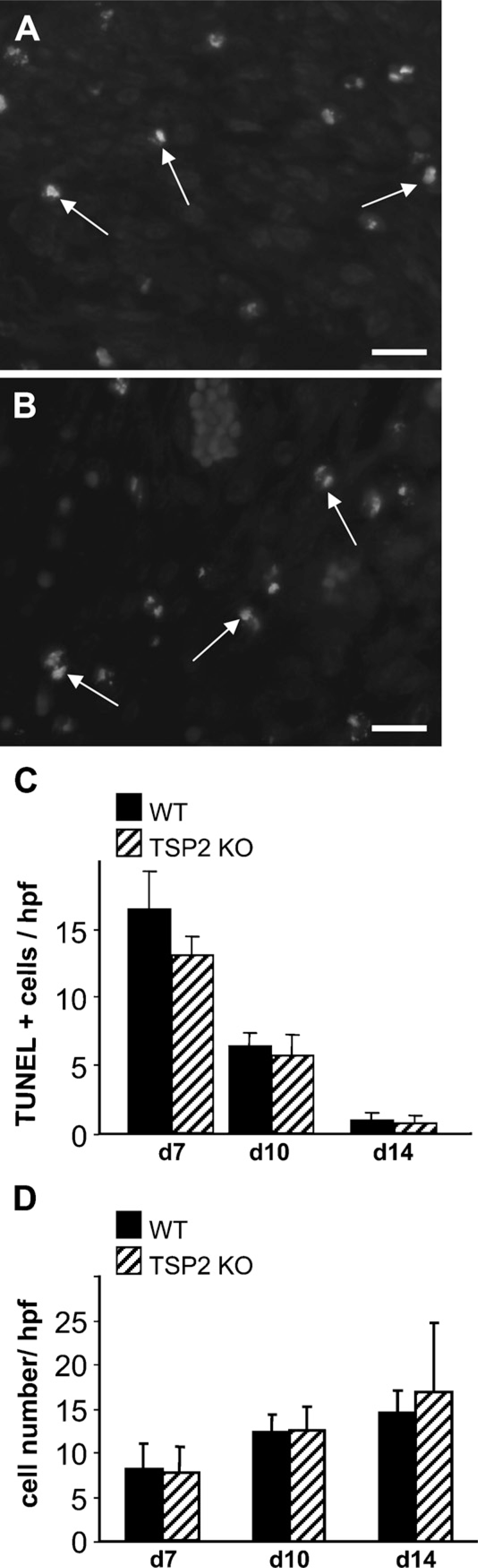
Equivalent apoptosis and proliferation in TSP2-null and WT wounds. Representative images from day 10 WT (
Increased MMP-2 and MMP-9 Levels in TSP2-null Wounds
The distribution of MMP-2 in day 10 excisional wounds from WT and TSP2-null mice was analyzed by immunohistochemistry (Figure 3). In the latter, a prominent association of MMP-2 immunoreactivity with the extracellular matrix could be observed. In contrast, the extracellular matrix of WT wounds showed a more limited distribution of MMP-2. To quantify these observations, day 7, day 10, and day 14 wounds were stained with anti-MMP-2 antibody and analyzed by histomorphometry. Consistent with the findings above, a large peak in MMP-2 levels in day 10 TSP2-null wounds was observed (Figure 3C). No differences in the levels of this MMP were observed in day 7 or day 14 wounds. The increase in MMP-2 coincided with the peak TSP2 expression in WT wounds (Kyriakides et al. 1999b; Agah et al. 2002). To confirm the semiquantitative histomorphometric analysis, day 10 wound extracts were subjected to zymographic analysis. Figure 3D (upper panel) shows the gelatinolytic activity of a 72-kDa protein and a 66-kDa protein, which were shown to be pro-MMP-2 and MMP-2, respectively, by Western blot analysis with an anti-MMP-2 antibody (lower panel). The presence of an additional pro-MMP-2 band, as seen in the Western blot, has been described previously (Haas et al. 1998), and is believed to be due to variation in glycosylation (Collier et al. 1988). Because MMP-2 is secreted as a complex with TIMP-2, we determined the levels of this inhibitor by Western blot and found them to be elevated in TSP2-null wounds (Figure 3D). Densitometric analysis of blots revealed a significant increase in the TIMP-2/β-actin ratio in TSP2-null wounds versus WT (1.0 ± 0.25 vs 0.07 ± 0.015;
An additional band, corresponding to a protein of ∼105 kDa in molecular mass, was observed in day 10 wound zymograms, and its levels were also increased in TSP2-null wounds (Figure 4D). This band was shown to correspond to pro-MMP-9 by Western blot analysis of the same extracts with an anti-MMP-9 antibody (Figure 4D). To examine the pattern of deposition of MMP-9, we performed immunohistochemical analyses of TSP2-null and WT wounds. Similar to the distribution of MMP-2, TSP2-null wounds displayed increased levels of MMP-9 in the extracellular space (Figures 4A and 4B). In contrast, WT wounds displayed higher levels of cell-associated MMP-9. In both groups, the deposition of MMP-9 was observed throughout the wound, including blood vessels and inflammatory cells. Histomorphometric analysis revealed an increased content of MMP-9 in day 10 TSP2-null wounds (Figure 4C). No significant differences were observed in day 7 and day 14 wounds. Western blot analysis of day 10 wound extracts for the presence of TIMP-1, an inhibitor that can complex with MMP-9, revealed elevated levels in TSP2-null wounds (Figure 4D). Similar to TIMP-2, densitometric analysis revealed a significant increase in the TIMP-1/β-actin ratio in TSP2-null wounds (2.0 ± 0.35 vs 0.1 ± 0.017;
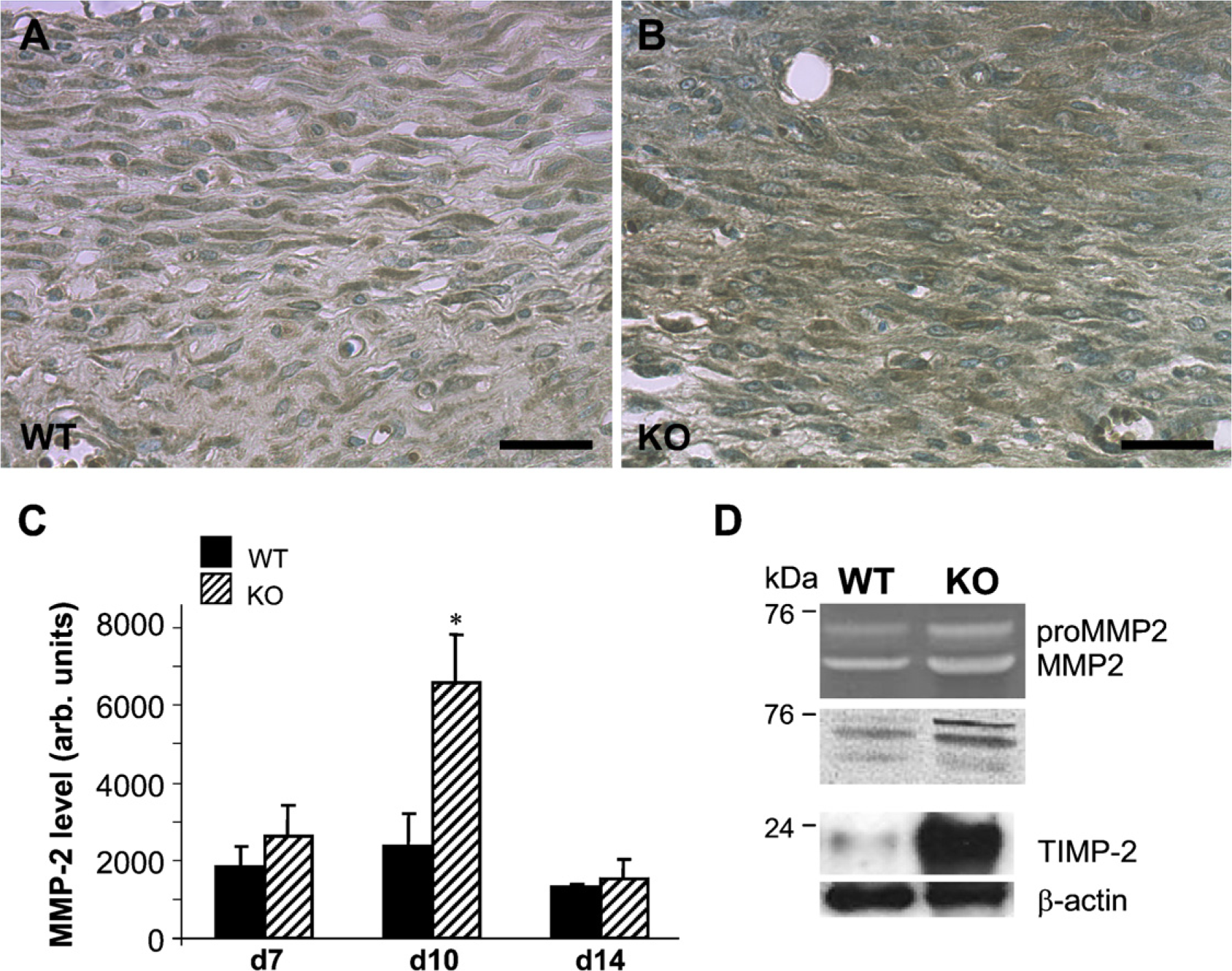
Increased MMP-2 in day 10 TSP2-null wounds. Representative sections from day 10 WT (
Elevated Levels of VEGF in TSP2-null Wounds
The increased deposition of MMP-2 and MMP-9 coupled with the increased angiogenesis in TSP2-null wounds prompted us to examine the levels of VEGF in wound extracts. Because both MMP-2 and MMP-9 have been shown to influence the availability of soluble VEGF, we performed a two-step extraction to recover soluble and insoluble VEGF. Western blot analysis of day 7, day 10, and day 14 wound extracts indicated increased VEGF levels in TSP2-null wounds relative to WT wounds (Figure 5). Both soluble and insoluble VEGF levels in control wounds decreased progressively from day 7 to day 14. In day 14 samples from WT wounds, mostly soluble VEGF was detectable. In contrast, VEGF levels in TSP2-null wounds were maximal in day 10 wounds and decreased by day 14, but remained high in comparison to control. The levels of soluble VEGF in day 10 wounds were also determined by ELISA and were found to be significantly higher in TSP2-null wound fluid (2173 ± 372 vs 448 ± 81 pg/mp;
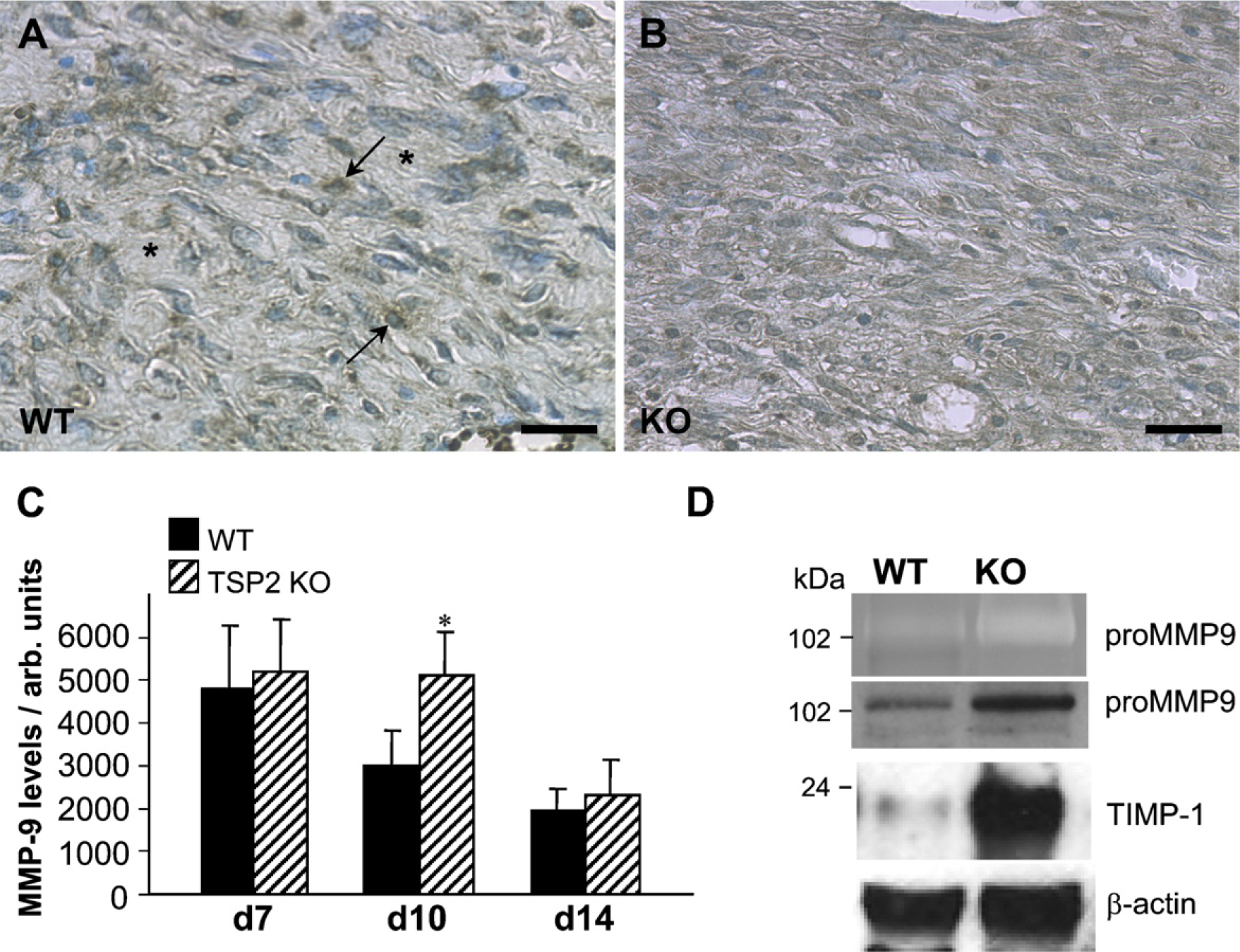
Increased MMP-9 in day 10 TSP2-null wounds. Representative sections from day 10 WT (
Normal Myofibroblast Differentiation in TSP2-null Wounds
Because of the reduced scarring of TSP2-null wounds, we analyzed sections by immunohistochemistry with an anti–smooth-muscle actin antibody to detect the presence of contractile cells such as myofibroblasts. WT wounds displayed numerous myofibroblasts and smooth-muscle cells/pericytes in day 7 and day 10 wounds (Figure 6). At day 10, the intensity of smooth-muscle actin immunoreactivity was increased, suggesting extensive fibroblast differentiation. Myofibroblasts were not evident in day 14 wounds, and only smooth-muscle cells associated with blood vessels could be observed. TSP2-null wounds displayed patterns of myofibroblast distribution that were indistinguishable from those in WT (Figures 6B, 6D, and 6F).
Abnormal Behavior of TSP2-null Wound Fibroblasts in Three-dimensional Collagen Gels
Collagen gel contraction assays are employed to simulate three-dimensional conditions in culture, and can evaluate cell behavior in either relaxed (released from the tissue culture plastic to contract) or stressed (maintained attached to the tissue culture plastic) matrices (Grinnell 2003; Rhee and Grinnell 2007). Fibroblasts isolated from TSP2-null wounds were compromised in their ability to contract relaxed collagen gels, displaying a 50% reduction in contractile ability compared with WT fibroblasts (Figure 7). This defect was evident in both the presence and absence of serum, but was more pronounced in the latter case. TSP2-null fibroblasts tended to form aggregations in relaxed gels and displayed aberrant cytoskeletal morphology in stressed gels. Specifically, in stressed gels, TSP2-null fibroblasts did not spread efficiently and assumed a bipolar morphology, which lacked the typical dendritic structures observed with WT fibroblasts (Figure 7B).
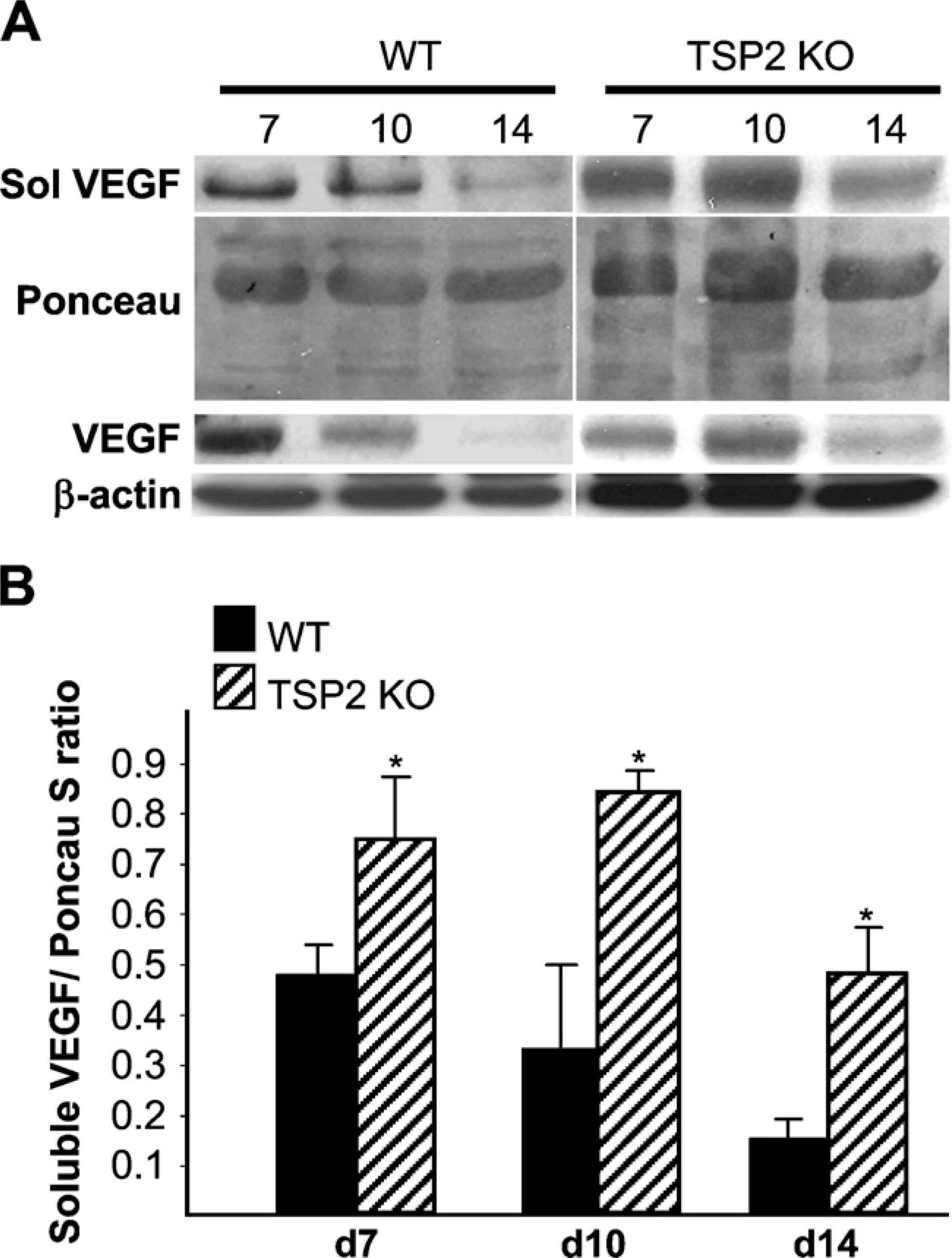
Increased levels of soluble vascular endothelial growth factor (VEGF) in TSP2-null wounds. (
Discussion
The wound-healing process is characterized by the sequential progression of overlapping phases that include hemostasis, inflammation, proliferation, and matrix remodeling (Singer and Clark 1999). The latter phase is carried out by a subset of MMPs (Moses et al. 1996; Parks 1999). Recent evidence suggests a role for matricellular proteins in the wound-healing process, in part through the regulation of the levels and perhaps activities of MMPs (Kyriakides and Bornstein 2003). Because TSP2-null mice have a wound-healing phenotype that can be attributed, in part, to elevated MMP-2 levels, we analyzed TSP2-null wounds for the presence of this MMP. In addition, we explored the two proposed mechanisms of inhibition of angiogenesis by TSP2, induction of apoptosis and cell cycle arrest, to see if either influenced wound neovascularization. We also analyzed the recovery of tensile strength in TSP2-null wounds to determine whether the cosmetic advantage of TSP2 deficiency, that is the reduced scarring in a TSP2-null environment, might be offset by a reduction in the mechanical integrity of the wounds. Finally, we analyzed the ability of TSP2-null wound fibroblasts to contract collagen gels.
Both TSP1 and TSP2 have been shown to be potent inhibitors of angiogenesis in vitro and in vivo (Bornstein 2001; Agah et al. 2002; Lawler 2002), and this inhibition is thought to result from both an increase in apoptosis and a reduction in cell cycle progression in microvascular ECs mediated via the interaction with CD36 and/or VLDL receptor (Jimenez et al. 2000; Nor et al. 2000; Armstrong et al. 2002; Oganesian et al. 2008). Analysis of TSP2-null and WT wounds by TUNEL staining revealed the presence of numerous cells undergoing apoptosis or necrosis, but the number of TUNEL-positive cells was not lower in TSP2-null than in WT mice. Detection of the cells in S-phase of the cell cycle with MIB-5 antibody also indicated that TSP2-null wounds contained the same number of proliferating cells as their WT counterparts. Both of these findings are surprising, because one would expect that in the absence of TSP2, the frequency of TUNEL-positive cells should be decreased, and that of proliferating cells should be increased, in comparison with control wounds. A possible explanation lies in the distinction between in vitro and in vivo systems. The ability of TSPs to increase apoptosis and reduce cell cycle progression was demonstrated in vitro in the presence of low serum levels (Jimenez et al. 2000; Armstrong et al. 2002; Oganesian et al. 2008). These conditions are not likely to reflect the wound environment, particularly in view of the high VEGF levels in both WT and TSP2-null wounds. Furthermore, the large number of inflammatory cells in wounds, which undoubtedly respond differently to TSPs than ECs and fibroblasts, are likely to mask the results of our experiments. However, it should be emphasized that TUNEL analysis of wound sections only detects cells retained within the wound. Therefore, it is possible that apoptotic/necrotic ECs are removed from the wound bed through the vascular system or by macrophages. Further, overall proliferation in WT wounds was greater at day 10 in comparison to day 7, suggesting that the elevated levels of TSP2 at the former time do not have a major influence on proliferation. In addition, the wound-healing phenotypes of TSP1-null and TSP2-null mice are opposite, with the former displaying delayed wound healing due to compromised inflammatory response. In fact, the ability of TSP1 or TSP2 to influence EC proliferation in vivo has not been reported. Thus, the involvement of a conserved TSP-specific anti-angiogenic mechanism in the development of these phenotypes is unlikely. Together, these results suggest that the observed high cellular content in TSP2-null wounds is maintained through increased recruitment, migration, or enhanced survival/retention of repair cells.
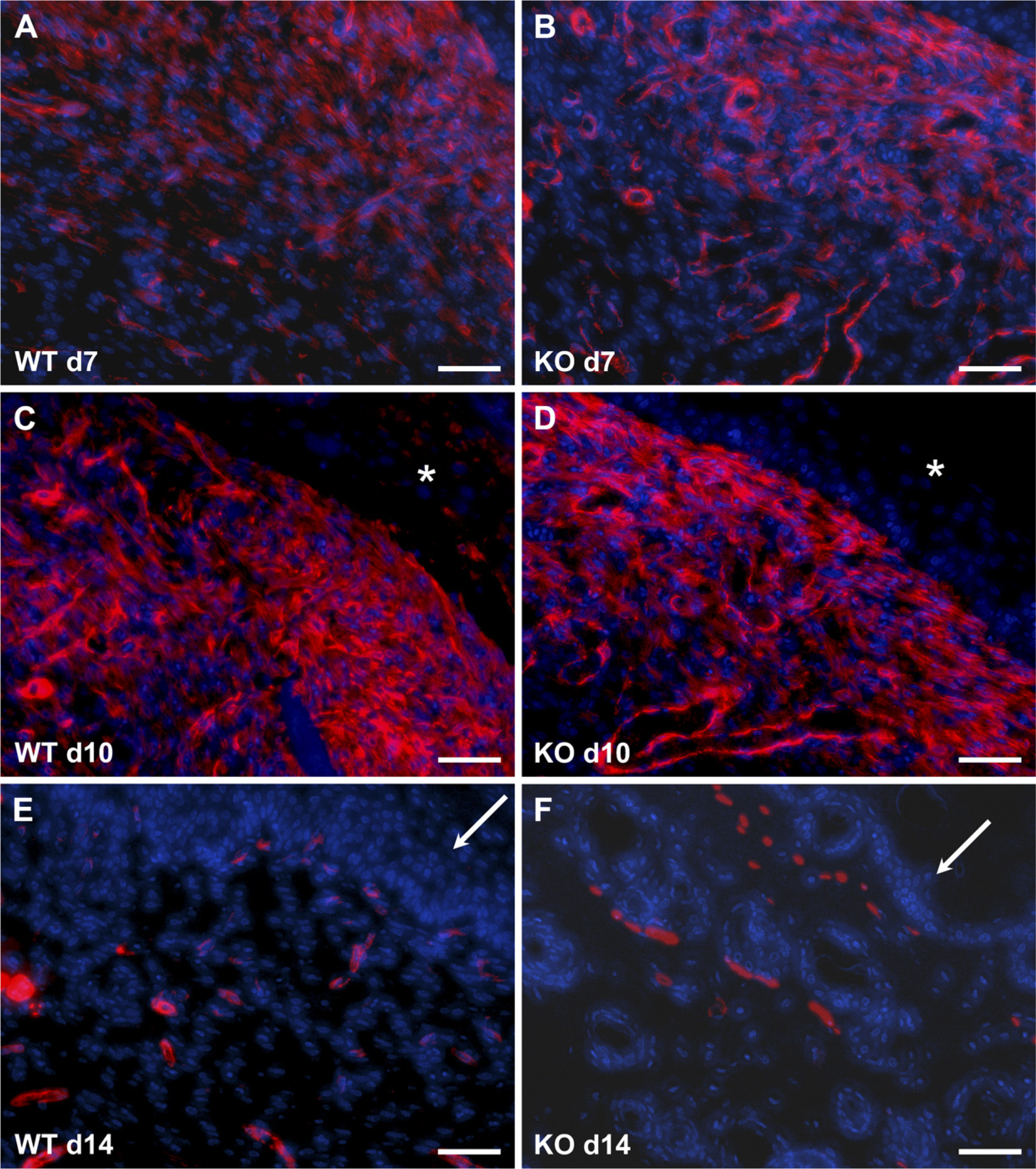
Normal myofibroblast content in TSP2-null wounds. Representative images of sections stained with anti–smooth-muscle actin antibody and visualized with fluorescence (red color) from WT (
Because TSP2 can regulate the levels of MMP-2 in dermal fibroblasts in vitro, we sought to determine whether this modulation was in effect during wound healing. Histomorphometry, zymography, and Western blotting confirmed that the levels of MMP-2 were increased in TSP2-null wounds at a time that coincided with the peak expression of TSP2 in WT wounds (day 10). Previously, we demonstrated that skin wounds in aged TSP2-null mice had increased levels of MMP-2 (Agah et al. 2004). This increase was manifest later, in day 14 wounds, which coincided with peak expression of TSP2 in age-matched WT wounds. Thus, there appears to be a strong inverse correlation between the peak TSP2 expression and MMP-2 levels. Interestingly, we recently showed that in hindlimb ischemia, the levels of MMP-9 but not MMP-2 are increased in the TSP2-null mice (Krady et al. 2008). In addition, zymography of wound lysates revealed increased levels and an additional band of gelatinolytic activity consistent with the molecular mass of MMP-9. Western blot confirmed the identity of the additional band as MMP-9, and histomorphometric analysis indicated increased MMP-9 levels in the wound bed of TSP2-null mice. It should be noted that the increased deposition of both MMP-2 and MMP-9 was observed predominantly in the ECM and is consistent with the concept that TSP2 mediates their clearance via catabolic pathways. In fact, the deposition of MMP-2 and MMP-9 in WT wounds was low in the ECM and high in cells. The presence of high MMP-9 levels was surprising, inasmuch as our previous in vitro studies of TSP2-null and WT dermal fibroblasts did not display an increase in MMP-9 (Yang et al. 2000). This finding can be explained by the fact that the majority of MMP-9 in the wound bed originates from macrophages that migrate into the wound. In addition, it has been previously reported that the levels of MMP-9 mRNA are highly induced in early murine wounds (days 1–7) but are not detectable in late wounds (day 13) (Madlener 1998; Madlener et al. 1998). Consistent with this study, we show a progressive decrease in the distribution of MMP-9 in WTwounds. It seems that TSP2 modulates MMP-2 and MMP-9 levels in vivo but in an injury- and age-dependent fashion, perhaps due to an altered injury cellular repertoire and MMP profile.
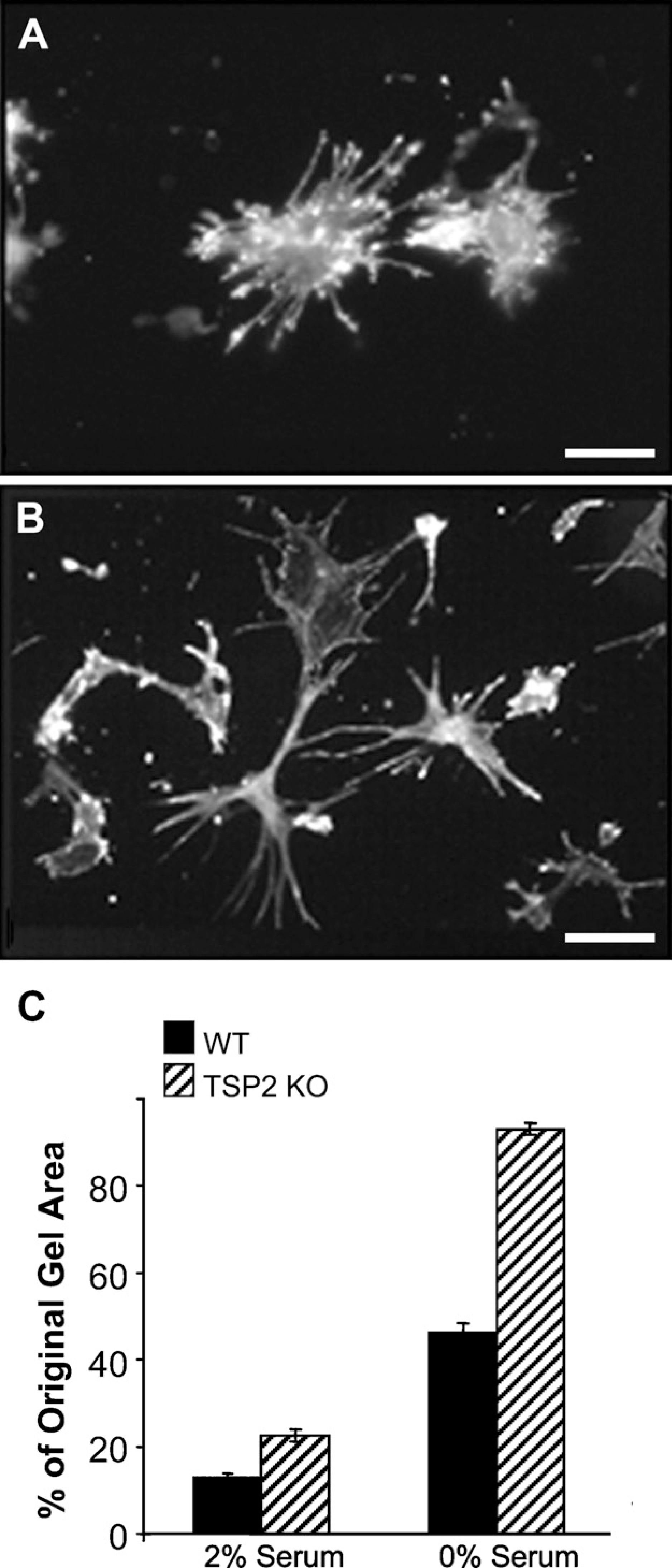
TSP2-null fibroblasts are compromised in their ability to contract collagen gels and display abnormal cytoskeletal shape. Cells suspended in stressed gels were visualized 24 hr following plating by staining with rhodamine-phalloidin. WT fibroblasts (
Because the expression and function of MMP-2 and MMP-9 are tightly linked to the expression of their respective inhibitors, TIMP-2 and TIMP-1, we evaluated TIMP expression in wound extracts. Previously, it was shown that the temporal expression of TIMP-1 and TIMP-2 mRNA in murine wounds mirrored that of MMP-9 and MMP-2, respectively (Madlener 1998; Madlener et al. 1998). We found low levels of TIMP-2 and TIMP-1 in day 10 WT wounds. In addition, higher levels of both TIMPs in TSP2-null wounds relative to WTwounds were observed, consistent with the fact that TIMPs can associate with MMPs during secretion (Overall and Lopez-Otin 2002). Concomitant increase in MMP-2 and MMP-9 with their respective inhibitors TIMP-1 and TIMP-2 seems contradictory to the proposed activation of MMPs indicated by increased soluble VEGF. This observation, however, is not unprecedented. MMP activity in vivo is regulated by zymogen activation, complex formation with the inhibitory TIMPs, or by compartmentalization (reviewed in Chakraborti et al. 2003; Ra and Parks 2007). In this study, the relative localization of TIMPs with their respective MMPs was not explored and might explain the observed increase in both MMPs and TIMPs.
Despite the increases in TIMP-1 and TIMP-2, we would expect extracellular free MMP-2 and MMP-9 to cause changes in matrix assembly, cell-matrix, and cell–cell interactions. The higher levels of MMP-2 and MMP-9 in the ECM of TSP2-null wounds suggest an intriguing scenario in which these two enzymes could influence both angiogenesis and matrix remodeling. Both processes have been shown to be dependent on the activity of MMPs (Werb et al. 1999; Heissig et al. 2003; Page-McCaw et al. 2007). In terms of altered ECM, we have demonstrated a reduction in the levels of isopeptide cross-links and tissue transglutaminase activity in TSP2-null wounds that are likely to alter collagen fibrillogenesis (Agah et al. 2005). Under these circumstances, it is reasonable to expect changes in the activation state of cytokines and growth factors (Werb et al. 1999; Heissig et al. 2003). Consistent with the ability of MMP-2 and MMP-9 to release matrix-bound VEGF, we detected elevated levels of soluble VEGF in TSP2-null wounds. Interestingly, it has been shown that increased levels of TIMP-1 can cause VEGF-induced neovascularization in the retina (Yamada et al. 2001). In addition, it is possible that TSP2 can reduce the levels of VEGF directly by an LRP-1-mediated clearance of a TSP2/VEGF complex, in a manner similar to that shown for VEGF and TSP1 in the ovary (Greenaway et al. 2007). The association of increased levels of MMP-9 with accelerated wound healing is also consistent with the observation that treatment of wounds with an MMP inhibitor retards healing (Agren et al. 2001; Mirastschijski et al. 2002).
Inhibition of TSP2 synthesis by localized gene delivery can reproduce the TSP2-null phenotype in WT mice by reducing scarring and accelerating healing (Kyriakides et al. 2002). Here we show that TSP2-null wounds recover tensile strength at a rate that is comparable to that of WT wounds. This finding is surprising, because uninjured TSP2-null skin is fragile and displays reduced strength (Kyriakides et al. 1998). The early phases of wound healing depend on rapid collagen biosynthesis and fibrillogenesis to replace the provisional matrix, but the organization of fibers in granulation tissue is unlike that of mature scar or normal skin. Even at 14 days postsurgery, wound strength is only a fraction of its ultimate level. We expect that longer-term observation of the properties of scars will probably reveal the defects in organization of collagen fibers that are characteristic of the TSP2-null phenotype.
The enhanced rate of healing of TSP2-null wounds also prompted us to utilize collagen gel contraction assays to explore the behavior of wound fibroblasts. Relaxed collagen matrices resemble the conditions found in dermis, and fibroblasts in this condition show reduced DNA and collagen synthesis, and reduced responses to growth factors (Grinnell 1994). To the contrary, stressed (attached) gels resemble wound granulation tissue conditions, and embedded cells continue to proliferate and secrete collagen (reviewed in Grinnell 1994). Ability to contract relaxed gels is commonly utilized to illustrate the underlying mechanism for alterations in wound contraction and scarring (Bullard et al. 1999a, b; Bradshaw et al. 2002). The fact that fibro-blasts isolated from TSP2-null wounds do not effectively contract collagen gels suggests that accelerated healing cannot be accounted for by enhanced contraction per se. Furthermore, we found that TSP2-null and WT wounds contained normal levels of myofibroblasts in day 7 and day 10 wounds, indicating that the isolated populations of wound fibroblasts used in the contraction experiments were comparable. Taken together, these observations suggest that the decrease in scarring in TSP2-null wounds could be due to reduced contractility of resident fibroblasts. Consistent with this suggestion, it has been shown in other mouse models that enhanced healing may not be accompanied by enhanced in vitro contraction (Ehrlich and Needle 1983; Bradshaw et al. 2002). Furthermore, the altered morphology of the TSP2-null fibroblasts in stressed collagen gels suggests that TSP2 functions primarily by modulating cell–matrix interactions.
In summary, the present study provides additional evidence for the modulation of MMP-2 and MMP-9 in skin wounds by TSP2 and implicates the TSP2–MMP–VEGF axis in the enhanced angiogenesis. Finally, our findings indicate that the mechanical integrity of healing TSP2-null wounds is adequate in the short term, and suggest that approaches that aim to improve healing by the transient inhibition of TSP2 will not compromise the integrity of the wound.
Footnotes
Acknowledgements
This study was supported by National Institutes of Health Grants AR-45418 (to PB), AG-06528 (to JMD), and GM-072194-01 (to TRK), the University of Washington Engineered Biomaterials Engineering Research Center (National Science Foundation grant EEC9529161) (to PB, TRK), and the Department of Veterans Affairs (to JMD).
We thank Jayasri DasGupta for performing the tensiometric analyses, Yumi Adachi and Emily Stainbrook for animal husbandry, and Matt J. Foster and Brett Schrom for technical assistance.