
Abstract
I wanted to be a cytochemist but encountered detours and then, in some of my work, became one of a different kind than classically defined. I recount this here to discourage young scientists from regarding cytochemistry as something that peaked in the past, but rather to be viewed as an entirely new form of the discipline, and so rich with opportunities. (J Histochem Cytochem 71: 475—480, 2023)
Keywords
Introduction
In my senior year at Syracuse University I took a course in cytology, the first semester general, the second cytochemistry (lecture and lab). I can’t say my intrigue with various dyes led to any epiphany as profound as Paul Ehrlich’s, who as a medical student famously had the insight that if cells and tissues take various dyes differently, their chemical composition must therefore differ. But I was fascinated by the different staining patterns, most notably how dyes known to be RNA-avid lit up the nucleolus, which was to become my career’s favorite organelle.
I stayed at Syracuse for my PhD and worked under the same professor who gave the cytology course, Seymour Gelfant. When I expressed my zeal for cytochemistry qua cytochemistry, he cautioned that a method doesn’t usually make a career and that innovations and advancements in methodology are ideally deployed to address a biological problem at hand. I had at this time also interacted with a card-carrying cytochemist, Alfredo Mariano Garcia at the nearby campus of the State University of New York Medical School. To my advisor’s point, Garcia was refining various cytochemical methods but for the purpose of optimizing their value in his broader work on lymphocyte cell biology.
I did two separate projects for my PhD thesis. One involved Feulgen cytophotometry to study the DNA contents of blood cells in the dogfish which, together with tritiated thymidine labeling, revealed that both circulating lymphocytes and red cell precursors are moving through the cell cycle, unlike the case for higher vertebrates. 1 In my collateral reading I became aware of how Feulgen cytophotometry had been so important in establishing the principal of DNA constancy, independently by Hans Ris at Rockefeller and Hewson Swift and Arthur Pollister at Columbia University, this being one of the key epistemological steps in the recognition that the gene is DNA.
I also read about the work of Zacharias Dische at Columbia, a biochemist not a cytochemist, who had developed a widely used colorimetric method for DNA in solution, based on diphenylamine. 2 This reaction involved the same acid hydrolysis of the N-glycosidic linkages of purines in DNA as does the Feulgen reaction and I got to wondering if diphenylamine might be used to detect RNA if the acid hydrolysis could be pushed to break the more stable RNA purine N-glycosidic linkages. This idea worked and led me to develop a method in which both DNA and RNA could be assayed in parallel, owing to the different spectra of the respective products. 3 Although this work was in the test tube, it further sensitized me to DNA and RNA colorimetric detection.
Cytochemistry and Histochemistry in the Early to Mid-20th Century
We might pause here for a moment to consider the field’s origins and where things stood at the time I was a student. Although there were even earlier developments, it was the German aniline dye industry that led to many of the stains employed as cytochemistry and histochemistry advanced. Among their importance were as tools for distinguishing cell types, of which the eponymous “eosinophil” is a familiar example. This archiving sometimes even involved extracellular staining, such as that of polysaccharides on the surfaces of some cells via the periodic acid Schiff reaction, also deployed for the detection of liver glycogen. We might also bear in mind that some “cytochemistry” didn’t even involve staining reactions. The most familiar example is ultraviolet cytophotometry, pioneered by Tjborn Caspersson in Stockholm, which revealed the high absorbance of the cytoplasm, a precursor to the recognition of RNA abundance in this compartment. And whereas optical microscopy was the theater of almost all cytochemistry and histochemistry at the time, there were important efforts at the ultrastructural level as well. Finally, there was the transformative idea of Albert Coons to use fluorescent antibodies as stains. Readers may be surprised how early this idea arose, 1942.
Detours Into Biochemistry and the Cell Biology of RNA
For the next 20 years my laboratory worked on RNA transcription, processing, and ribonucleoprotein (RNP) assembly both biochemically and to some extent in living cells. 4 In 1989, a graduate student in my lab, Jin Wang, had the idea to try doing actual cytochemistry in living cells. Instead of using a cytochemical stain to detect an endogenous molecule in the cell, he decided to dye-label that molecule in the test tube and then microinject it, as a tracer. The idea worked perfectly for pre-mRNA in the nucleus, 5 and later a postdoc Marty Jacobson extended this approach for several nucleolar RNAs,6,7 whereby their dynamic movements could be tracked (Fig. 1). 8 Anent my professor’s advice, this live cell cytochemistry approach did lead to a biological discovery. With this method we demonstrated that the nucleus is the site of assembly of the signal recognition particle, 9 the first case of realizing that this organelle is doing something beyond building ribosomes (and we now know it is doing even more.)
Rapid localization of RNase P RNA in the nucleolus. RNase P RNA was transcribed in vitro, coupled with rhodamine, microneedle injected together with fluorescein-labeled dextran into the nucleus of a normal rat kidney (NRK) cell and imaged 4 min later. This rapid localization of a known nucleolar RNA reinforced our belief that “fluorescent RNA cytochemistry” would open doors to investigating the intranuclear dynamics of various other RNAs.6,7,9–11 Scale bar = 5 µm. Reproduced from Jacobson et al. 8 by permission of the Company of Biologists.
Could Nucleic Hybridization in Live Cells Be Deployed as “Cytochemistry?”
Just when my laboratory seemed to be leaving live cell experiments for more and more in vitro molecular biology I was joined by a postdoc, Joan Politz, who daringly wanted to pursue the use of nucleic acid hybridization to track RNA in living cells. Her strategy was to tag oligodeoxynucleotides with caged fluorescein so that once hybridization had occurred in the cell, the bound oligos could be photoactivated. She deployed this method to label nascent ribosomes in the nucleolus of living cells and then, by uncaging the hybridized oligos could track the emergence of the ribosomes from the nucleolus into the nucleoplasm and cytoplasm (Fig. 2). 10 In other applications we tracked the movements of poly(A) RNA within the nucleus, including it dynamic shuttling between nucleoplasm and nuclear speckles, the sites of mRNA splicing. 11 The only limitation in this method is having a pinhole in the microscope light path so as to precisely direct the photoactivation to the desired site. Most laboratories who do imaging have this ability.

Tracking nascent ribosomes in the nucleus. Rat L6 myoblasts were exposed for one hr to a set of oligodeoxynucleotides complementary to 28S ribosomal RNA, each of which was conjugated with caged fluorescein. The fluorescein was uncaged at a nucleolus (circle in second panel) and the movement of the nascent 60S ribosomes was tracked at 110 msec, 1.4 sec, 3.6 sec, and 26 sec (next four panels). The 60S ribosomes depart the nucleolus and progressively distribute throughout the nucleoplasm. That many remain in the nucleolus after 26 sec is compatible with the known overall timescale of 60S ribosome synthesis, with the observed departure representing the most mature 60S ribosomes at the time of uncaging. At later time points they began to appear in the cytoplasm (not shown).
Another Lapse but With a Cytochemist’s Subconscious Mind
After the forgoing work my laboratory pursued other problems in cell biology, especially on the plurifunctionality of the nucleolus but without any obvious entry points for the live cell cytochemistry approach. Then, in 2015 we got the idea that the emerging clustered regularly interspaced short palindromic repeats (CRISPR) technology could enable our work on chromosome dynamics. This effort was led by a postdoc, Hanhui Ma. The idea was to use a nuclease-inactive form of the CRISPR Cas9 protein, labeled with green fluorescent protein (GFP), together with an expressed guide RNA to reach the target. This was conceptualized before us by another group and their elegant publication encouraged us. 12 Over the next few years we refined this method as to spectral range and sensitivity (Fig. 3).13–17 And again, to my professor’s advice, we did it apply it to biological problems. In one case we were able to actually measure the CRISPR on-target residence time 14 and in another we defined the dynamic range of mobility of chromosomes in the interphase nucleus, 17 confirming and extending a study during my postdoc in which I had used 3 H-actinomycin as a “cytochemical” to initially reveal this. 18 In the most recent refinements of our CRISPR-based genome-labeling methods we have been able to observe the process of heterochromatin formation in living cells. 19
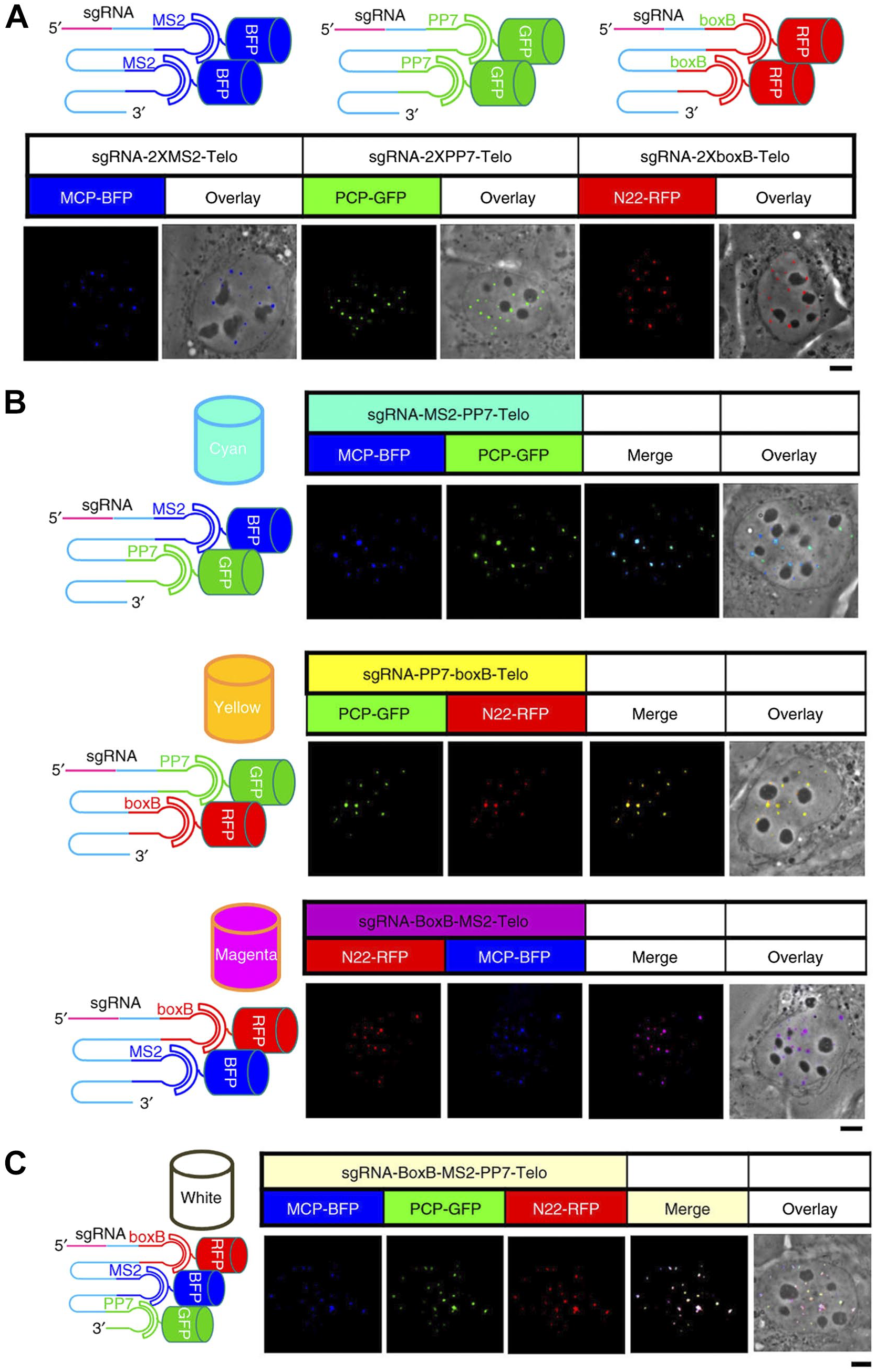
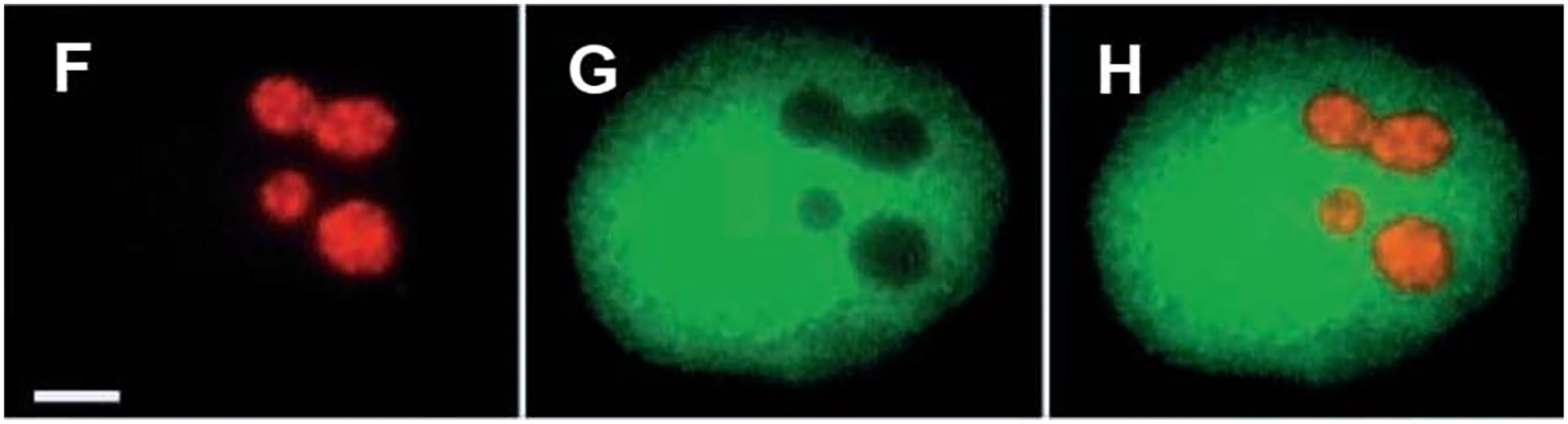
“CRISPRainbow,” an iteration of our initial genome-labeling methods.13,14 The strategy here was to link the guide RNAs (targeting telomeres in this case) with 3 scaffolds consisting of pairs of aptameric RNA sequences specific for attachment of various colored proteins. This binary scheme is not the color combinations we learned as children (e.g., blue + yellow = green) but is enabled by the spectral filters in our microscope, yielding three “primary” colors (blue, green, and red, Panel A) and four “secondary” colors (cyan, yellow, magenta, and white, Panels B and C; I ask readers to forgive me for calling white a color but it was useful to have in our deployment of this system.) In these various dopings the guide RNAs retained their faithful targeting of the target telomeric sequences (Panel B). This was a far stretch from my earliest ideas of becoming a cytochemist. Reproduced from Ma et al. 15 by permission of Nature Springer Publishing Group. Abbreviations: BFP, blue fluorescent protein; GFP, green fluorescent protein; RFP, red fluorescent protein; MCP, bacteriophage MS2 coat protein; PCP, Pseudomonas bacteriophage 7 coat protein; N22, bacteriophage N22; PP7, Pseudomonas phage 7.
Cytochemistry Today
Cytochemistry is a field defined by methodology. We can be reminded by Sydney Brenner’s dictum “Progress in science depends on new techniques, new discoveries, and new ideas, probably in that order.” “Cytochemistry today” also intersects with chemical biology as applied to live cells, as was pioneered by the late Roger Tsien and others. 20 Revolutionary subsequent advances by Frances Arnold, Carolyn Bertouzzi, Linda Hsieh-Wilson, Alice Ting, Xiaowei Zhuang, and others are enormous opportunities, just as was the initial introduction of GFP. 21 The revered Yale X-ray crystallographer Tom Steitz was known for asking lab members: “Did you solve your structure today?” I would like to ask today’s students: “Have you envisioned a place for today’s cytochemistry in your research?”
Footnotes
Acknowledgements
In our deployment of caged fluorescein, we were aided by the generous provision of this reagent by Timothy Mitchison (Harvard Medical School), synthesized by himself and not yet commercially available.
Competing Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
The author conceived of and wrote the article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The author’s research described in this article was funded by the U.S. National Institutes of Health, the U.S. National Science Foundation and the Human Frontier Science Program Organization.