
Abstract
Renal tissue injury initiates inflammatory and fibrotic processes that occur to promote regeneration and repair. After renal injury, damaged tissue releases cytokines and chemokines, which stimulate activation and infiltration of inflammatory cells to the kidney. Normal tissue repair processes occur simultaneously with activation of myofibroblasts, collagen deposition, and wound healing responses; however, prolonged activation of pro-inflammatory and pro-fibrotic cell types causes excess extracellular matrix deposition. This review focuses on the physiological and pathophysiological roles of specialized cell types, cytokines/chemokines, and growth factors, and their implications in recovery or exacerbation of acute kidney injury.
Keywords
Introduction
Acute kidney injury (AKI) is a multiphasic disease that represents a global concern. The onset of AKI increases the risk of developing chronic kidney disease (CKD) or end-stage renal disease (ESRD). Furthermore, CKD affects up to 16% of the global population and pre-existing CKD significantly increases the risk of new AKI episodes.1–3
As a response to renal injury, inflammatory pathways are initiated, cytokines and chemokines are secreted, reparative processes are launched, and pro-fibrotic cells are activated. This controlled response offers regeneration of damaged tissue, while incomplete or persistent signaling from inflammatory and fibrogenic cells can result in fibrosis 4 (Fig. 1). Interestingly, incomplete repair can be dormant and re-initiate upon an insult, such as AKI. 5 Appreciating AKI and CKD as “interconnected syndromes” 3 and understanding the molecular mechanisms and cellular crosstalk during injury will elucidate pathways for targeted intervention. It is important to note that there are many extensive pathways and mechanisms that play significant roles in renal inflammation and fibrosis, such as hypoxia, autophagy, and metabolism; however, only select molecules and processes are described in this review.
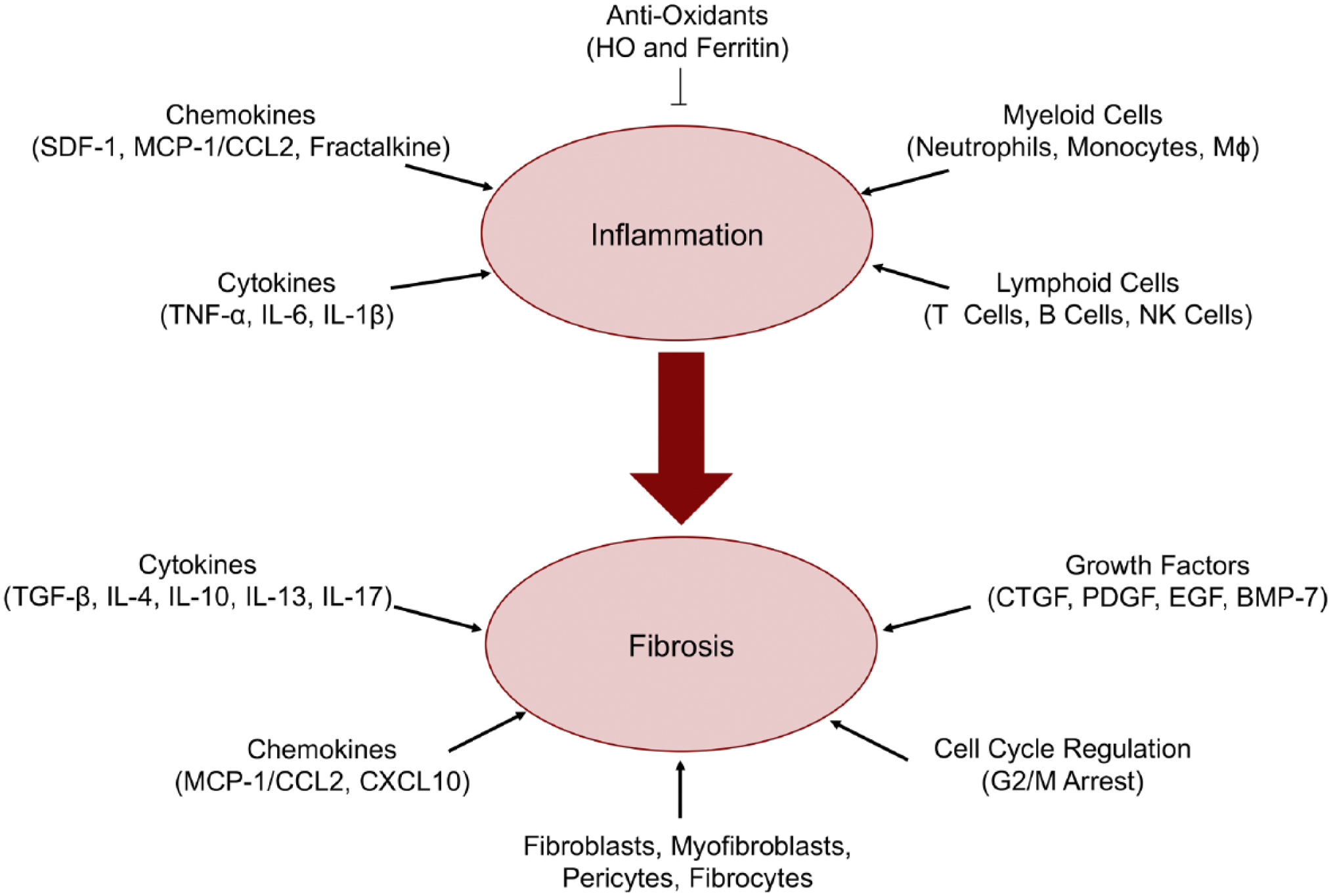
Distinct cell types, signaling proteins, and growth factors contribute to renal inflammation and fibrosis. Renal injury induces inflammation, which drives fibrosis. Coordination of a multitude of cell types, cytokines and chemokines, antioxidants, growth factors, and regulatory mechanisms modulates these responses. Meticulous control of these factors can drive repair of damaged tissue; however, if dysregulated, injury is exacerbated. Abbreviations: Mɸ, macrophage; NK cells, natural killer cells; TNF-α, tumor necrosis factor-α; IL, interleukin; SDF-1, stromal cell-derived factor-1; MCP-1, monocyte chemoattractant protein-1; CCL2, chemokine (C-C motif) ligand 2; HO-1, heme oxygenase-1; CTGF, connective tissue growth factor; PDGF, platelet-derived growth factor; EGF, epidermal growth factor; BMP-7, bone morphogenic protein-7; CXCL10, C-X-C motif chemokine 10; TGF-β, transforming growth factor-β.
Inflammation
Early inflammation is characterized by the presence of neutrophils and macrophages, recruited and activated via cytokine release in damaged tissue, which in turn stimulate the adaptive immune response 6 (Fig. 2). A time-dependent release of pro-inflammatory mediators in the early injury stage is relieved by anti-inflammatory factors secreted by recruited and resident cell populations, 7 resulting in injury resolution and healing; however, abnormal and persistent inflammation coupled with protracted release of factors, such as transforming growth factor-β (TGF-β), causes maladaptive repair processes and progressive renal disease 8 (Fig. 2).
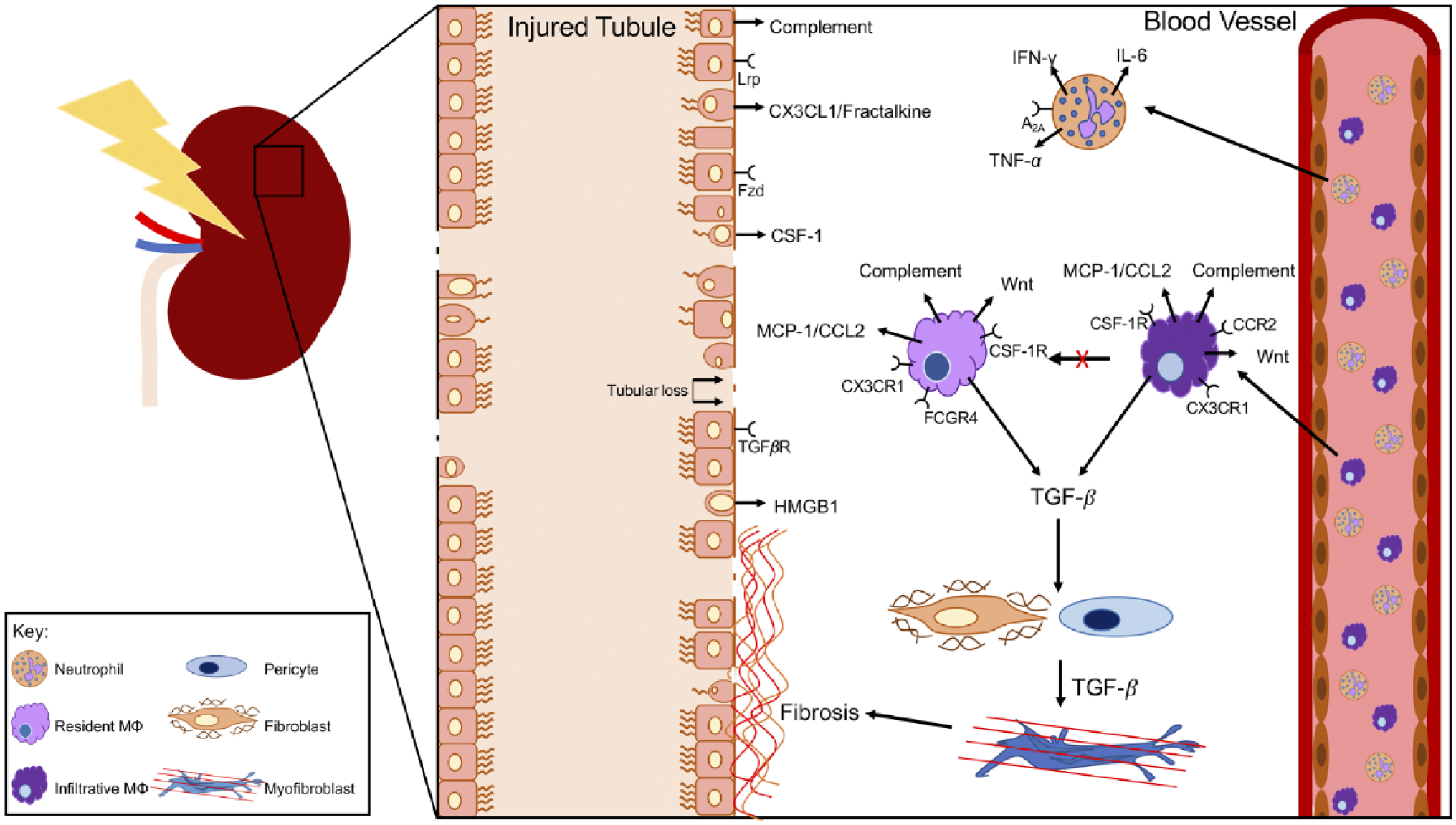
Neutrophil and macrophage interaction with injured kidney epithelium and interstitial cells. Neutrophils infiltrate into the injured tissue within hours of the event and express pro-inflammatory cytokines and enzymes for degrading extracellular matrix, providing space for activity of macrophages in clearance of debris. Infiltrative macrophages migrate from the blood stream into the kidney interstitium and are short-lived. Their numbers often peak within 1 to 3 days after injury and decline with resolution of acute inflammation. Few of these infiltrative macrophages differentiate into tissue resident macrophages. Infiltrative macrophages express chemokine and growth factor receptors, including CX3CR1, CCR2, and colony stimulating factor-1 receptor (CSF-1R). They express secreted proteins, including transforming growth factor-β (TGF-β), monocyte chemoattractant protein-1 (MCP-1/CCL2), Wnt ligands, and complement proteins. Tissue resident macrophages, in contrast to infiltrative, renew by in situ proliferation. Their numbers remain relatively constant after ischemia-reperfusion–acute kidney injury (IR-AKI). They express receptors including CX3CR1, CSF-1R, and Fc γ receptor 4 (FCGR4). They express secreted proteins including TGF-β, CCL2, Wnt ligands, and complement proteins. Certain Wnt ligands are differentially expressed by kidney resident macrophages compared with infiltrative. For example, Wnt4 is expressed at higher levels by resident macrophages. TGF-β secreted by both infiltrative and tissue resident macrophages can be trophic for fibroblast to myofibroblast differentiation. Myofibroblasts are critical pro-fibrotic cells in the kidney interstitium that secrete collagens and ECM proteins that promote interstitial fibrosis. Epithelial cell death and damage results in expression of complement proteins, CX3CL1 (fractalkine), CSF-1, and high mobility group box-1 (HMGB1). Each of these molecules are recognized by and can activate leukocytes. Also, the epithelium expresses receptors for signaling molecules produced by leukocytes, including Fzd/Lrp (Wnt ligand receptors), and TGF-βR. Fzd, frizzled (Wnt receptor); Lrp, low density lipoprotein receptor (Wnt co-receptor); IFN-γ, interferon-γ; Wnt, wingless-related integration site.
Mediators
Cytokines
Cytokines are produced predominantly by inflammatory cells,9,10 but their expression is also observed in epithelial cells and interstitial cells.11,12 Pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-8 (IL-8) are associated in humans with worsened acute outcomes, 13 chronic inflammation, and CKD. 14
Colony Stimulating Factor-1 (CSF-1)
CSF-1 is a cytokine known to modulate the severity of AKI in animal models,15–17 by mediating signaling pathways and promoting recovery after ischemia-reperfusion (IR) through activation of pro-healing macrophages.15,17 Classically, CSF-1 is known to promote macrophage differentiation from bone marrow precursors.18,19 Tissue resident macrophages and bone marrow-derived mononuclear phagocytes, suggested to be of independent cellular origin, 20 may both produce and respond to CSF-1. 15 Mononuclear phagocytes are a group of cell types including monocytes, macrophages, and dendritic cells (DCs). Renal tubules express CSF-1R, 16 which suggests its role as a pro-healing signal in animal models of AKI. 15 Proximal tubules (PTs) are also a large source of CSF-1 and when PT-specific CSF-1 is deleted, macrophages and DCs are polarized to a pro-inflammatory phenotype and fibrosis is exacerbated. 17
TNF -α
TNF-α is expressed mainly by macrophages, renal tubular cells, and mesangial cells and regulates damage, promoting inflammation and cell death signaling pathways, including death receptor and caspase-mediated cell death 21 and those mediated by c-Jun/AP-1 22 and NF-кB.5,23–25 TNF-α is also upregulated during healing and has inflammation-resolving effects via TNFR2.26,27 A proposed mechanism by which TNF-α activates NF-κB, promoting cell survival, was demonstrated in a study by Micheau and Tschopp. 28 Serum levels of TNF-α may be useful as an early biomarker to predict severity of AKI. 29 An animal study of the AKI to CKD transition suggests TNF-α promotes persistence of damage promoting M1-like macrophages and augments progression to interstitial fibrosis. 30 TNF-α is known to play a major role in renal inflammation and fibrosis, acting as a stimulant for the release of monocyte chemoattractant protein-1 (MCP-1/CCL2), interleukin-1β, and TGF-β1 in glomerular disease. Although, it has also been suggested that soluble TNF receptors may neutralize excess TNF-α and thereby weaken the inflammatory response. 31
TGF -β
Acting mainly on fibroblasts, mesangial cells, and tubular cells, TGF-β exists in three isoforms (TGF-β1, TGF-β2, and TGF-β3) and is commonly regarded for its role in fibrosis. 5 TGF-β is expressed and secreted by many cell types during injury.32,33 TGF-β additionally plays important roles in embryonic development, stem cell differentiation, immune cell signaling, and tissue repair. 34 TGF-β expression is particularly abundant in renal tissue, as well as in monocyte and macrophage populations, lymphocytes, and platelets. 35 In renal pathophysiology, TGF-β expression drives extracellular matrix (ECM) remodeling and expansion of pro-fibrotic cell types such as fibroblasts and myofibroblasts.36,37 In fact, TGF-β overexpression under quiescent conditions results in renal tubular cell autophagy and subsequent fibrosis. 38 Furthermore, in a model of murine post-contrast AKI (PC-AKI), it was determined that TGF-β/Smad3 signaling is activated upon injury, leading to increased Ctgf, Mmp-9, and Collagen IVa gene expression. In addition, mice with PC-AKI experience increased apoptotic cell death with an accompanying decrease in proliferation. 39
AKI in mice causes upregulation of TGF-β signaling, and PT-specific deletion of the TGF-β receptor II (TGFβRII) reduces injury and apoptosis. 40 However, a paradoxical effect was observed in a murine CKD model, where PT-deletion of TGFβRII led to increased tubulointerstitial fibrosis, while also impairing Wnt/β-catenin signaling. In fact, artificial stimulation of Wnt/β-catenin signaling reduced tubular G2/M cell cycle arrest and apoptotic cell death, 41 suggesting the importance of TGF-β-Wnt/β-catenin crosstalk during renal injury. Similarly, myeloid-specific TGFβRII deficiency in mice caused a pro-inflammatory macrophage phenotype, decreased renal TGF-β expression and macrophage-specific TGF-β signaling, suppressed renal macrophage infiltration, and attenuated fibrosis in AKI. 42 Extremely complex in nature, TGF-β activity can be either beneficial or detrimental depending on disease state. Therefore, the benefits of therapeutic intervention via TGF-β for controlling inflammation and fibrosis remains to be seen. 5
Interleukins
IL-1β serves as a pro-inflammatory signal that may increase damage after injury and increase the risk of interstitial fibrosis. Administration of IL-1β, both in vitro and in vivo increased expression of NGAL (neutrophil gelatinase-associated lipocalin), a biomarker for kidney injury. 43 In IR-induced AKI to CKD transition in rats, IL-1β and IL-18 remained elevated after TNF-α and macrophage numbers returned to baseline levels. Mitochondrial protection in this model demonstrated an augmentation of IL-1β and IL-18 levels, which was associated with improved fibrosis. 44 Furthermore, IL-1β, in concert with IL-4 and platelet-derived growth factor (PDGF), is involved in fibrocyte differentiation. 45 IL-1 receptor associated kinase-M (IRAK-M) deletion promoted renal atrophy, atubular glomeruli formation, and interstitial fibrosis. IRAK-M served as an inhibitory signal for IL-1β-mediated signaling in macrophages, indicating that IL-1β activated macrophages toward a pro-inflammatory phenotype augmenting tissue damage and fibrosis. 30 IL-33 is another interleukin that acts as an “alarmin,” released from cells as a response to stress or necrosis and is thought to cause sustained inflammation. 5 In addition, IL-33, has been associated with renal inflammation and fibrosis following unilateral ureteral obstruction (UUO) 46 and promoted fibrosis following IR-AKI. 47
IL-10 is primarily produced by mesangial cells in normal renal physiology and has been found to be either protective or detrimental, depending on renal disease and disease state. 5 Administration of IL-10 reduced serum IL-6, kidney collagen III, and kidney α-SMA expression in murine IR. 48 Other studies have utilized IL-10 expression as a biomarker for reduced inflammation.49,50 In UUO, IL-10 protected from interstitial fibrosis. 51 However, it has been suggested that IL-10 and TGF-β can together cause pro-inflammatory cascades. Furthermore, IL-10 is responsible for the secretion of Cystatin-C, a cysteine protease inhibitor, that is currently used diagnostically to detect renal function. 5
IL-22 is a cytokine in the same family as IL-10, produced by Th17, Th22, and innate lymphoid cells. 52 Both in vitro and in vivo, IL-22 promoted epithelial integrity after injury. 53 TLR4-mediated activation of interstitial leukocytes may induce IL-22 expression in these immune cells, and IL-22 promoted tubular epithelial healing after AKI. 54 Furthermore, it was recently found that IL-22 treatment activated STAT2 and AKT pathways, thereby attenuating injury after renal IR. 52 The role of anti-inflammatory cytokines is important as, whether acute or chronic, they may reduce the risk of subsequent interstitial fibrosis. 55
Chemokines and Damage Patterns
MCP-1, also called CCL2, serves to recruit mononuclear phagocytes from the vasculature into injured tissue. CCR2, the receptor for MCP-1/CCL2, is expressed by monocyte precursors and is required for their emigration from the bone marrow. In humans, MCP-1/CCR2 is associated with decreased capillary density, increased macrophage infiltration, and functional CKD. 56 In addition, expression of CCR2 by intravascular mononuclear phagocytes promotes renal infiltration in the presence of CCL2. 57 Conditional overexpression of kidney injury molecule-1 by renal epithelium in a mouse model resulted in increased MCP-1 expression and subsequent macrophage trafficking in the kidney. 58 In a model of mouse UUO, gene transfer of an MCP-1 N-terminal deletion mutant decreased CCR2-positive macrophage infiltration in the kidney. Furthermore, such mutants experienced significantly lesser TGF-β expression and interstitial fibrosis than untreated controls, suggesting a role for MCP-1 in development of renal fibrosis in CKD. 59
CX3CR1 is expressed by mononuclear phagocytes in the kidney in mice and humans. When the ligand CX3CL1 (also called fractalkine) binds to CX3CR1, activation of inflammatory cells occurs. Kidney macrophages promote interstitial fibrosis after injury due to production of pro-fibrotic molecules, such as TGF- β and PDGF-B. 60 For example, in an animal model of folic acid nephropathy, CX3CL1 expression was associated with interstitial fibrosis. 61 In patients with chronic renal allograft rejection, expression of both CX3CL1 and CX3CR1 were observed in the tubulointerstitium and epithelial cell basolateral membrane. 62
Complement is associated with both antibody-mediated and non-antibody-mediated kidney diseases. 63 Recently, Thorenz demonstrated that complement 5a receptor 2 (C5aR2)-deficient mice demonstrated protection from inflammatory tissue damage and fibrosis after IR-AKI. 49 Furthermore, cyclosporine nephrotoxicity has been associated with complement activation in the tubulointerstitium. 64 In a mouse kidney transplantation model where Crry-deficient donor kidneys were transplanted into complement receptor or wild-type recipients, C3aR deficiency in recipients protected from increases in inflammatory cell numbers and tubulointerstitial injury. 65
Sphingosine-1-phosphate (S1P) is recognized by G protein-coupled receptors expressed on the surface of the mouse renal endothelium secondary to AKI. Deletion of sphingosine-1-phosphate receptor, S1PR1, aggravated inflammation and fibrosis after AKI. 66 In a cell-based therapy approach in mice, a therapeutic benefit was observed from adoptively transferred S1pr3-deficient DCs following AKI. 67 These studies support the hypothesis that S1P serves as a signal that, when combined with DCs are able to bind S1P, promotes inflammatory damage after AKI, potentially further leading to the development of fibrosis and ESRD.
High mobility group box-1 (HMGB1) serves as a damage pattern actively released by mononuclear phagocytes and passively released by necrotic cells during tissue injury. 68 The HMGB1 ligand can be bound by toll-like receptors (TLR) and cause downstream activation of macrophages. 69 Serum from AKI patients showed increased levels of HMGB1, implying it may be a useful biomarker. 70 Leemans and colleagues associated upregulation of TLR2, a receptor for HMGB1, and HMGB1 upregulation with UUO in mice. 71 Furthermore, Tian and colleagues demonstrated that HMGB1 from both macrophages and tubular cells polarized macrophages to a pro-inflammatory phenotype, while inhibiting HMGB1 release mitigated fibrosis in a model of UUO. 72 This suggests HMGB1 expression, when dysregulated, can augment inflammatory response and exacerbate fibrosis in CKD.
Antioxidants
Reactive oxygen species (ROS) play important roles in hormone synthesis and signaling, cell proliferation, bacterial defense, and activation of various ion channels and receptor signaling. However, dysregulation of ROS exacerbates renal inflammation and fibrosis. 73 To combat oxidative stress-induced injury, there are several endogenous antioxidants in place to mitigate such damage: heme oxygenase (HO), ferritin, and superoxide dismutase, catalase, and others.
HO
During oxidative injury, heme is destabilized from proteins (e.g., hemoglobin, myoglobin, cytochromes), causes ROS production, and protein and lipid oxidation. To this effect, HO catabolizes heme to generate carbon monoxide, biliverdin, and iron, the latter of which is sequestered by ferritin, discussed later in this review. HO-1, the more widely studied, inducible isoform, is relatively low in abundance in quiescence, and is upregulated and protective in AKI.74–78
A seminal study performed by Nath and colleagues in 1992 demonstrated that treatment with an HO inhibitor aggravated renal function in a rat model of rhabdomyolysis. However, with induction of HO-1 by treatment with hemoglobin, these deleterious effects were attenuated. 77 Hull and colleagues used myeloid-specific deletion of HO-1 and reported impaired recovery and fibrosis after bilateral IR-AKI. 79 Similarly, Bolisetty and colleagues deleted HO-1 from the PTs and found worsened renal structure and function and exacerbated apoptotic cell death after cisplatin nephrotoxicity. This injury was circumvented by selective overexpression of PT-HO-1. 76 Taken together, these studies suggest that therapeutic intervention by means of HO-1 stimulation could protect against inflammation and fibrosis in renal disease.
Ferritin
Ferritin is an iron storage protein made up of two distinct subunits: heavy chain (FtH) and light chain (FtL), capable of sequestering iron. 80 Although FtL is responsible for electron transfer, FtH has been more extensively studied in the context of renal injury, as it confers ferroxidase activity. Although FtH is historically thought of as a cytosolic iron regulatory protein, new studies detected its presence in the mitochondria and nucleus,81,82 as well as identifying its role in osteoblastic differentiation of vascular smooth muscle cells, 83 angiogenesis, 84 inflammation, 85 and fibrosis. 86
Ferritin is upregulated as a response to pro-inflammatory cytokines, termed inflammation-induced hyperferritinemia, which is different than classical iron-dependent ferritin expression. In fact, studies show that specific cytokines such as IL-1β and TNF-α stimulate FtL and FtH synthesis.87,88 Rogers demonstrated that human hepatoma cells increase translation of FtH and FtL after stimulation with IL-1β. 87 In addition, FtH was found to be induced by TNF-α, acting directly on its transcription as a response to oxidative stress. 88
Furthermore, Zarjou and colleagues found that when FtH is deleted from the PTs, mice experience worsened renal function, significant damage to the renal architecture, and exaggerated apoptosis in models of cisplatin nephrotoxicity and rhabdomyolysis. 89 In related studies, FtH was shown to modulate inflammation in models of AKI 86 and sepsis. 85 Further studies should be conducted to manipulate the mechanisms of both FtH and FtL in mitigating renal injury for the development of targeted therapeutics.
Cells
Myeloid Cells
Myeloid lineage immune cells that are observed in the post-AKI setting and participate in renal inflammation and fibrosis consist of neutrophils (polymorphonuclear cells), monocyte/macrophages, and DCs. Monocytes are intravascular myeloid lineage cells that have the potential to differentiate into macrophages or DCs.
Neutrophils
Neutrophils are acute responders, demonstrating increased numbers within 2 hr after renal IR injury. 90 They produce enzymes which degrade ECM. Therefore, if neutrophil recruitment continues past the acute phase, the damage they inflict upon tissues may promote fibrosis. For example, in a recent study using the unilateral IR-AKI to CKD animal model, neutrophil numbers were found to be increased up to 2 weeks after the initial insult, and these mice demonstrated notable interstitial fibrosis. 91 Taken together, as neutrophil depletion or inhibition of neutrophil accumulation prevents AKI, 92 these data suggest that neutrophil manipulation in renal injury could substantially modulate disease.
Macrophages and DCs
Mononuclear phagocyte subtypes present in the mouse kidney in the quiescent state and in AKI models have been reviewed previously. 93 A non-human primate study was conducted demonstrating that infiltrative CD11b-expressing myeloid cells promote interstitial fibrosis after AKI. 94
Controversy exists regarding identifying macrophages versus DCs in the kidney. Regardless of their precise identity, there are a substantial number of CX3CR1-expressing mononuclear phagocytes distributed throughout the healthy and inflamed kidney.57,95–98 Stamatiades identified F4/80Hi-expressing cells as predominantly macrophages due to their sessile state, endocytic function, and expression of MerTK, FCGR1, and FCGR4,96,99 and these conclusions have been confirmed by subsequent studies.20,98
Macrophages were proposed to produce Wnt7b in a post-injury response that promoted tissue regeneration. 100 However, it is evident that Wnt7b is not the only Wnt ligand produced by myeloid inflammatory cells in the kidney. Importantly, the receptors for Wnt ligands are expressed by the renal parenchyma and by macrophages themselves. 100 Using β-catenin/TCF/LEF reporter mice, extensive activation of the canonical Wnt pathway in the renal parenchyma was observed in IR injury. 100 We recently found that Wnt4 was the most highly expressed Wnt ligand by kidney resident macrophages 6 days after AKI. 20
DCs represent a rare cell type in the healthy kidney and are characterized by the F4/80LowCD11cHi surface phenotype, MHCII expression in naive DC, and demonstrate expression of transcription factors Zbtb46 and Batf3.101,102 Evidence suggests an important role for intrarenal DC in glomerulonephritis. 102
Eosinophils
Peripheral eosinophilia is a hallmark observed in acute interstitial nephritis (AIN) in humans, while local renal inflammatory infiltrates contain a mixture of cells including eosinophils, macrophages, and lymphocytes. 103 In drug-induced AIN, if the offending agent is not removed, the disease can be associated with subsequent interstitial fibrosis. Thus, eosinophils should be considered in translational studies of renal inflammation and fibrosis.
Lymphoid Cells
Lymphoid lineage immune cells are involved in renal inflammation and fibrosis, including T cells, B cells, and natural killer (NK) cells. In animal models of AKI, the AKI to CKD transition, or kidney fibrosis models, T cells, B cells, and NK cells are observed.91,104–106 Lymphoid follicle-like structures form within the kidney as tertiary lymphoid tissues in severe AKI to CKD animal models.107,108 Although B cells and NK cells have been observed in these models, their function in renal inflammation and fibrosis remain poorly understood.
T Cells
The predominant role of T cells in renal inflammation and fibrosis is in chronic inflammation and coincides with continual macrophage activation. T cells polarized to the Th2 phenotype induce pro-fibrotic alternative activation of macrophages and may promote kidney fibrosis. 109 However, in a study of omeprazole-induced AIN, Th17 and Th1 cells were predominately observed. 110 This indicates a specific type of insult may polarize T cells, through which Th1, Th2, or Th17 cells may promote fibrosis.
Animal models of AKI indicate T cells participate in early pathophysiology following AKI.111,112 RAG1−/− mice, which are deficient in T cells and B cells, subjected to IR-AKI, are not protected from injury compared with wild-type controls at early time-points through 72 hr post-IR. 113 Acute lymphocytic infiltration may be important in renal inflammation and fibrosis due to increased severity of initial injury and its association with severity of eventual organ fibrosis.
T regulatory cell (Treg) numbers increase after ischemic-preconditioning in the post-AKI setting in animal models.114–116 Due to their function in dampening and resolving immune responses to tissue injury, it follows that Tregs may play an important role in determining outcomes in renal inflammation and fibrosis.
Tissue Regeneration Versus Fibrosis
Damaged cells release factors that initiate inflammatory responses, stimulating temporary wound healing and ECM deposition (Table 1). Vascular permeability and vasodilation accompany this response, coupled to secretion of matrix metalloproteases from surrounding cells, allowing for entry of inflammatory mediators and cellular motility. 117 The first responders, mainly neutrophils and macrophages, enter the site of injury to clear debris and secrete cytokines and chemokines, which aid in activating surrounding cells. They also elicit new blood and lymphatic vasculature to further aid in injury clearance.6,118 Pro-fibrotic factors can be secreted by infiltrating leukocytes, which activate pro-fibrotic cells to differentiate into myofibroblasts, which line, seal, and contract the site of injury (Fig. 3). In adaptive repair, these cells work together with epithelial and endothelial cells to regenerate the damaged tissue. However, in the setting of an unrestrained inflammatory response, permanent scar formation can occur. 117 Coordinated orchestration of inflammatory cells, secreted cytokines and chemokines, pro-fibrotic factors and cell types is necessary to assure healing takes place instead of fibrosis and renal dysfunction.
Selected Contributors to Renal Inflammation and Fibrosis.
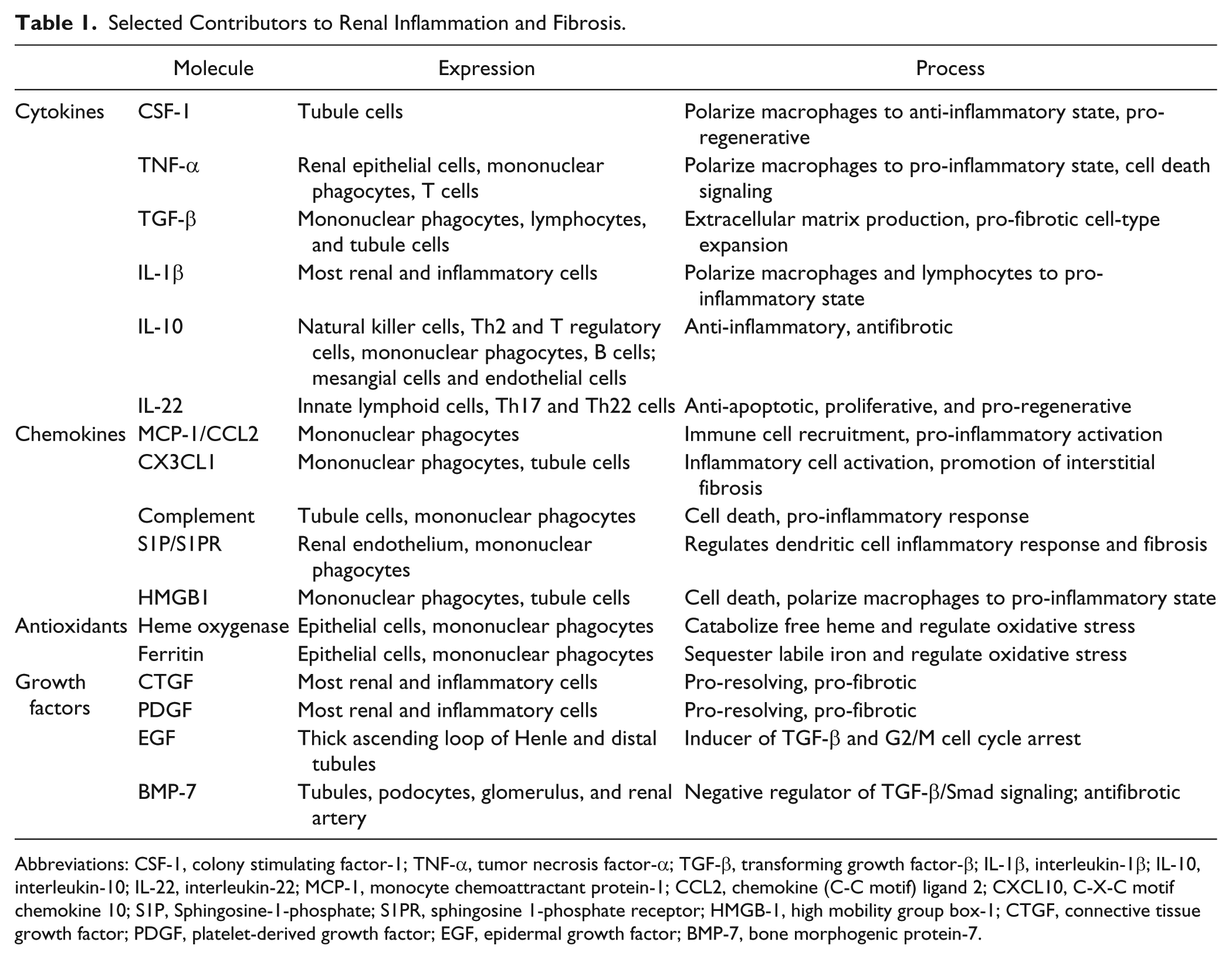
Abbreviations: CSF-1, colony stimulating factor-1; TNF-α, tumor necrosis factor-α; TGF-β, transforming growth factor-β; IL-1β, interleukin-1β; IL-10, interleukin-10; IL-22, interleukin-22; MCP-1, monocyte chemoattractant protein-1; CCL2, chemokine (C-C motif) ligand 2; CXCL10, C-X-C motif chemokine 10; S1P, Sphingosine-1-phosphate; S1PR, sphingosine 1-phosphate receptor; HMGB-1, high mobility group box-1; CTGF, connective tissue growth factor; PDGF, platelet-derived growth factor; EGF, epidermal growth factor; BMP-7, bone morphogenic protein-7.
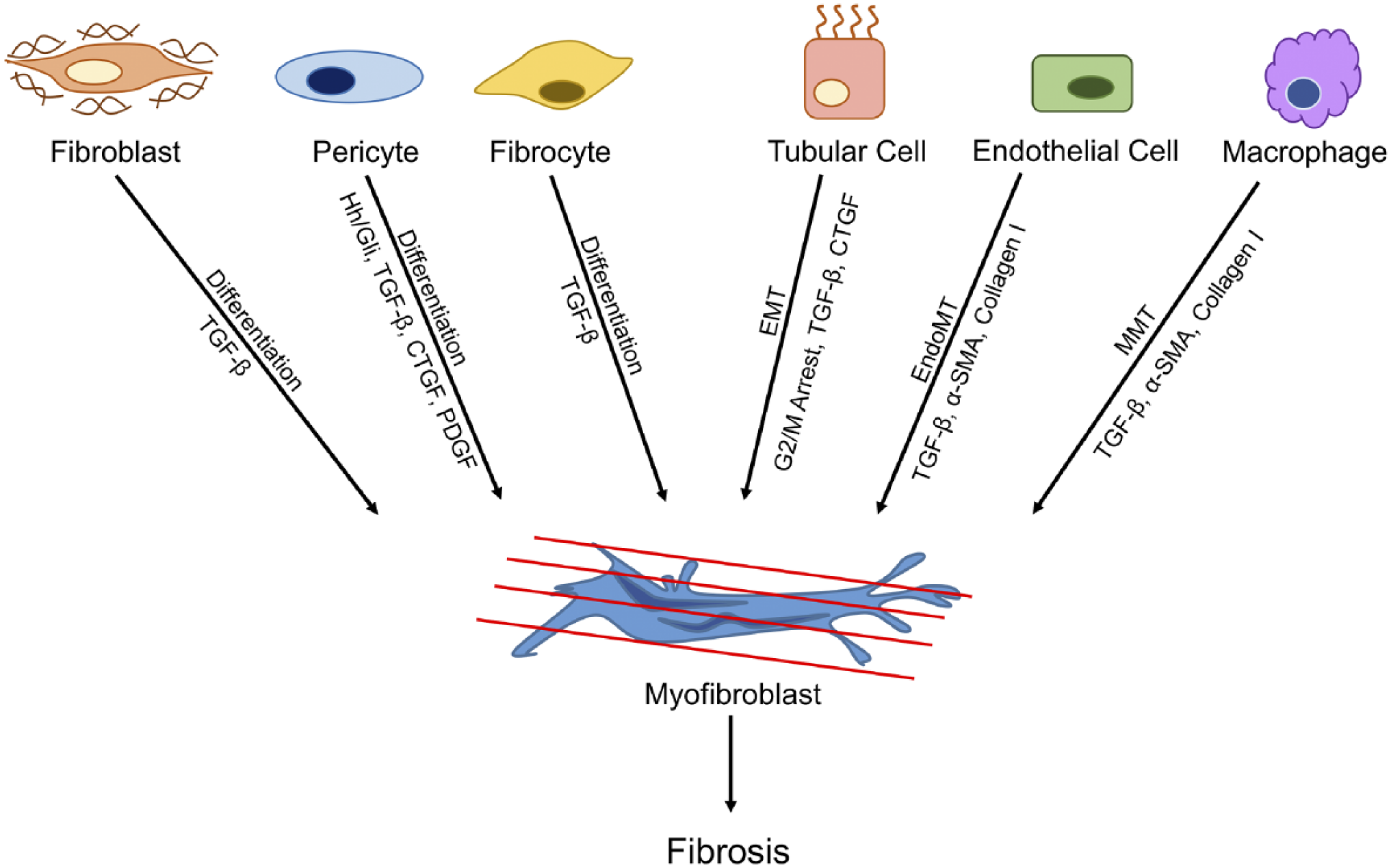
Origins of myofibroblasts in renal fibrosis. Many renal cell types can differentiate into myofibroblasts during fibrosis in response to various stimuli, including fibroblasts, pericytes, fibrocytes, endothelial cells, macrophages, and tubular cells. Abbreviations: TGF-β, transforming growth factor-β; Hh/Gli, Hedgehog/GLI signaling pathway; CTGF, connective tissue growth factor; PDGF, platelet-derived growth factor; EndoMT, endothelial-to-mesenchymal transition; MMT, macrophage-to-myofibroblast transition; EMT, epithelial-to-mesenchymal transition; α-SMA, α-smooth muscle actin.
Mediators
Connective Tissue Growth Factor (CTGF)
CTGF is induced by TGF-β, secreted by fibroblasts and is implicated in fibrosis by transducing signals through Smad, ERK, and Wnt signaling pathways. Physiologically, CTGF plays critical roles in nephron development and glomerular epithelial cell differentiation.5,119 In injury, CTGF is regulated by both TNF-α and TGF-β 120,121 and can, in turn, induce TNF-α, IL-6, NF-κB, and MAPK signaling.122,123 CTGF is regarded as a pro-fibrotic protein; fibroblast-specific overexpression of CTGF causes systemic fibrosis, affecting the kidney, vasculature, skin, and lungs. 124 In addition, treating mesangial cells with recombinant human CTGF induces collagen I and fibronectin, as well as auto-expression. 125 This induction was reversed and fibrosis was attenuated in vivo by use of CTGF antisense oligonucleotides in a model of rat UUO. 126 Manipulation of CTGF could be a potential therapeutic avenue against renal fibrosis. In fact, recent studies attribute the antifibrotic and antiproliferative effects of rapamycin to reduced CTGF expression. 127
PDGF
PDGF is regarded for organogenesis of the kidneys, brain, lungs, and vasculature, as well as physiological and pathophysiological processes. 128 PDGF exists in four major isoforms, PDGF-A, PDGF-B, PDGF-C, and PDGF-D, and has two receptors, PDGFR-α and PDGFR-β, which are present on most cells in the kidney. 129 While PDGFR is expressed by mesangial cells, fibroblasts, and vascular smooth muscle cells in the kidney, renal epithelial cells (podocytes, tubular cells, etc.) do not express PDGFR either in the normal nor in the injured state. 129
PDGF-D overexpression in mice caused mesangio-proliferative glomerulonephritis and tubulointerstitial injury. 130 In both human and mouse tissue, PDGF-D and its receptor, PDGFR-B, are upregulated on mesenchymal cells, and injured tubular cells express PDGF-D. Mice deficient in PDGF-D had significantly less fibrosis in murine UUO and unilateral IR, while mice that overexpressed PDGF-D had exacerbated renal fibrosis. 131 In addition, fibrocyte differentiation into myofibroblasts is regulated in part by PDGF, as well as IL-4 and IL-13, 45 further framing it as an active player in the development of organ fibrosis.
Epidermal Growth Factor (EGF)
EGF is responsible for cell survival, proliferation, metabolism, and differentiation. 132 EGF receptor (EGFR) expression is seen particularly in proximal and distal tubules, while the ligand is expressed by thick ascending loop and distal tubules. 133 EGFR is partially responsible for the final number of nephrons and collecting duct morphology. 134 Furthermore, EGF plays a pivotal role in kidney development, whereas its deletion caused incomplete collecting duct development and premature death. 135 In disease, EGF signaling induces TGF-β, 136 G2/M cell cycle arrest, 137 and pro-inflammatory factor release 138 as a response to injury.
Studies by Humes and colleagues revealed that controlled EGF signaling plays a significant role in repair after renal IR in rats. This study demonstrated the important physiological role of EGF after AKI, accelerating tubular repair. 139 Clinically, urinary excretion of EGF is used as a biomarker which helps detect injury in renal disease. 133 On a cellular level, EGFR contributes to fibrosis by binding several pro-fibrotic ligands aside from EGF, including transforming growth factor-α (TGF-α), heparin-binding EGF-like growth factor (HB-EGF), amphiregulin, betacellulin, and epiregualtin. 137
Persistent EGF activation in pathological settings has been associated with accelerated renal failure. Using transgenic mice that selectively express HB-EGF in the PT, Overstreet et al. demonstrated tubular dysfunction and fibrosis. The authors additionally blocked EGFR activity, which caused attenuation of fibrosis. They postulated that persistent PT-EGF expression stimulated cell cycle arrest and dedifferentiation of tubular cells. 140 Contrarily, studies have shown that EGFR inhibition delayed recovery in murine renal IR as well as folic acid nephrotoxicity, suggesting an imperative role early in AKI.141,142
Bone Morphogenic Protein-7 (BMP-7)
BMP7 is involved in both kidney development and injury response, which acts against TGF-β induced EMT by induction of E-cadherin. 143 BMP7 plays a large role in development, as seen in seminal studies in which global BMP7 deficiency impaired kidney development.144,145 BMP7 was also involved in metanephric mesenchymal survival by blunting cell death pathways. 146 In the adult kidney, BMP7 was expressed in the tubules, podocytes, glomerulus, and renal artery, 147 while its receptor, BMP7-RII, was expressed in the glomerulus, PTs, and collecting duct. 148
In UUO, Sun and colleagues studied transcriptional and translational dynamics in BMP7 and found that there was more BMP7 mRNA present than BMP7 protein. There was also increased levels of miR-384-5p, a microRNA that decreases BMP7 expression. Suppressing miR-384-5p activity lessened the severity of renal injury in their model. 149 Administration of recombinant human BMP7 to mice caused greater SMAD1/5/8 phosphorylation, lesser collagen expression and deposition, and attenuated SMAD3 and Akt signaling. 150 In fact, AA123, a small molecule that mimics BMP-7 activity has shown promise in attenuating murine renal fibrosis, while its structural analog is currently being investigated in a phase II clinical trial. 5
Cells
Fibroblasts
Fibroblasts exist in quiescence within the renal interstitial potential space, playing a crucial role in maintaining structure of the kidney. After injury, fibroblasts are activated by various stimuli to repair damaged tissue. However, persistent inflammation and cytokine release instigates anomalous fibroblast activation, leading to α-smooth muscle actin (α-SMA) expression, typical of myofibroblast activation, and excessive production of collagen I/III/IV and fibronectin.
The role of fibroblasts in both AKI (folic acid nephrotoxicity) and CKD (UUO) have been explored. 151 Studies showed that prominent fibroblast-specific gene expression patterns in AKI were different than those in CKD, modulating disease outcomes. Induction of Wnt signaling pathways was observed, with an increase in Wnt4 and Wnt5a. Authors suggest that Wnt signaling derived from fibroblasts inhibited repair processes and augmented the pro-inflammatory response. 151 Prostaglandin E receptor 3 (PTGER3) aids in repair by preventing fibroblast activation in addition to being negatively regulated by TGF-β. Levels of PTGER3 are reduced in UUO, suggesting attenuation of fibroblast activity due to TGF-β signaling. These results indicate that recovery from renal injury depends on suppression of fibroblasts, activation of ECM remodeling, and an inflammatory response. 151
Fibroblasts are a highly dynamic and plastic cell type, changing role and activation state depending on location and disease state. 152 Recent studies indicate that a cell type switch of tubular cells to fibroblasts occurs in renal injury but can also be reversed. Using specific transcription factors (Emx2, Hnf1b, Hnf4a, and Pax8), mouse and human fibroblasts can be re-differentiated into induced renal tubule cells, which not only share the same expression profile, and morphological and functional characteristics but are also able to amalgamate into tubular structures in decellularized kidney scaffolds. 153 Taken together, studies indicate that pharmacological manipulation of fibroblast differentiation could be monumental in preventing fibrosis in renal disease.
Pericytes
Intertwined around the renal microvasculature, pericytes play important physiological roles in development, angiogenesis, maturation of vessels, immune surveillance, and injury response. In pathological processes, pericytes are regarded as playing a major part in the development of renal fibrosis. Pericytes are myofibroblast progenitor cells154,155 and have been shown to undergo pericyte to myofibroblast transition under the direction of the Hedgehog/GLI, TGF-β, PDGF, and CTGF pathways. 156
Fibrotic remodeling that occurs in the glomerular region, predominantly driven by collagen I/IV and fibronectin, disrupts normal filtration and blood flow, while fibrosis that occurs between the tubules and capillary system, driven by α-SMA, can affect cellular transport processes and waste removal. 157 In fact, kinetic remodeling and microscopy over the course of UUO revealed that pericytes differentiated into myofibroblasts and contributed to fibrosis, a process most likely initiated by vascular injury. 155
Furthermore, Xavier et al. 158 demonstrated a more complex role of pericytes and their relationship with immune cells during renal injury and fibrosis. Murine UUO and folic acid nephrotoxicity demonstrated the ability of pericytes to secrete C1q, a protein complex involved in complement activation. Xavier et al. found that this causes a cascade of events, such as pro-inflammatory cytokine expression, Wnt/β-catenin signaling, and collagen production. Deletion of C1q did not ameliorate renal fibrosis after UUO. However, global C3 deficient mice experienced decreased renal macrophage infiltration and subsequent fibrosis. 158
Fibrocytes
Fibrocytes are derived from CD14+ bone marrow monocytes, differentiated by way of PDGF, IL-4, IL-13, and TGF-β,45,159 and are key players in fibrosis for their ability to produce collagen. 160 Fibrocytes also expressed markers for both hematopoietic cells and stromal cells and are able to differentiate further into myofibroblasts upon TGF-β stimulation.160–162 Pilling and colleagues 160 described the presence of cell-type specific markers, CD45, 25F9, and S100A8/A9 in fibrocytes, but not in monocytes, macrophages, and fibroblasts.
Sakai and colleagues found fibrocytes infiltrated the damaged kidney in UUO, which paralleled the gradual development of fibrosis. 163 Furthermore, extra-renal fibrocytes were able to migrate into the injured kidney, reliant on CCR2 expression. 164 Sakai et al. 165 also identified that blockage of angiotensin II type-1 receptor (AT2R) signaling reduced the number of fibrocytes in the bone marrow and infiltrating into the kidney, suggesting a role for AT2R in fibrocyte activation and accumulation.
Myofibroblasts
As terminally differentiated cells, myofibroblasts are crucial in pathological ECM, fibronectin, and collagen production, and are mainly located in the interstitium of the kidney. Growth factors, such as TGF-β, fibroblast growth factor, TNF-α, and IL-1, stimulate pericytes154,166 and fibroblasts to differentiate into these cells. 152 In elegant studies performed by Humphreys and colleagues, fate tracing revealed that pericytes instead of epithelial cells were the source of myofibroblasts. Furthermore, they suggested that endothelial disruption may induce fibrosis due to the communication that occurs between endothelial cells and pericytes via factors such as PDGF. 154
Kramann and colleagues studied the hedgehog (Hh) pathway, specifically the role of myofibroblast-specific GLI1 and GLI2, in the development of renal fibrosis in UUO. Interestingly, GLI2 knockout mice experienced reduced fibrosis due to cell cycle arrest in myofibroblasts. This was corroborated in vitro, where arsenic darinaparsin induced GLI1 and GLI2 expression and subsequent cell cycle arrest in a 10T1/2 cell line, effects that were reversed by overexpression of GLI2. Administration of a GLI antagonist (GANT61) after UUO also confirmed this result, halting myofibroblast cell cycle progression and reducing fibrosis. 167
Mechanisms of Cellular Transdifferentiation
Cell Cycle Arrest
In cellular homeostasis, renal tubular cells divide with carefully maintained cell cycle progression 168 to combat normal tubule loss. 169 Cell cycle regulation is crucial in renal physiology. For example, G1 cell cycle arrest during injury protects damaged cells from replicating damaged DNA; however, if the cell cycle remains arrested, cell senescence occurs. 170
Interestingly, PT cells that express vimentin, CD24, and CD133 can reversibly dedifferentiate to help repair damaged epithelial cells. 171 These cells were able to undergo clonal expansion, driving re-establishment of tubular function. 172 Injection of CD24+CD133+ PT cells after murine AKI stimulated renal engraftment of PTs, thereby augmenting renal function. 173 Studies further validated that mature epithelial cells were responsible for promoting tubular repair, not intratubular stem cell or progenitor cell populations. 174
During adaptive repair, renal epithelial cells utilize cell cycle entry to regenerate the damaged nephron, driven by expression of cell cycle regulatory proteins (e.g., p53, p21, and p16).175–178 However, G2/M cell cycle arrest after AKI led to fibrosis, as tubules produced an excess of pro-fibrotic factors such as CTGF and TGF-β (Fig. 3). 171 Using IR-AKI, aristolochic acid nephrotoxicity, and UUO, it was demonstrated that tubules that were arrested in the G2/M phase produce pro-fibrotic factors TGF-β and CTGF, which were induced secondary to JNK signaling. Tubular fibrotic factor release was ameliorated by inhibiting the ataxia telangiectasia mutated pathway, JNK signaling or, in unilateral AKI models, by contralateral nephrectomy. 179 The cell cycle protein, p21, has been implicated in nephrotoxic models of AKI as well. Cyclin inhibitor p21 is induced after cisplatin nephrotoxicity and its inhibition caused worsened structural damage and significant mortality. Mice deficient in p21 demonstrated tubular cell advancement to S phase and necrosis, while damaged cells in wild-type controls did not undergo cell cycle progression.180,181
Epithelial-To-Mesenchymal Transition (EMT)
EMT occurs first in development, when epithelial cells differentiate into mesenchymal cells and vice versa until development is complete (Fig. 3). 182 Epithelial cells can dedifferentiate into mesenchymal cells secondary to injury, during which transitioning cells migrate from the basement membrane to the interstitial space and differentiate into fibroblasts. 182 In the kidney, this process is driven by TGF-β. Phosphorylation of Smad3 allows it to complex with Smad4 and translocate to the nucleus to activate target pro-fibrotic genes, causing tubule cells to dedifferentiate into mesenchymal cells and driving development of renal fibrosis. 183
It was thought that fibroblasts active during tissue injury are derived from EMT 184 ; however, Humphreys and colleagues published evidence that myofibroblasts are largely derived from resident pericyte/perivascular fibroblasts in the kidney. 154 Damaged cells do in fact play an important role in activation of myofibroblasts and also possess pliant qualities, such that they are able to dedifferentiate after damage into mesenchymal-like cells or return to an epithelial cell state. 185 Human tubular epithelial cells from biopsies demonstrated changes suggestive of the switch to a mesenchymal-like state. 186
Endothelial-To-Mesenchymal Transition (EndoMT)
EndoMT is a process during which endothelial cells dedifferentiate, lose cell-specific markers, and develop into mesenchymal or myofibroblast cells (Fig. 3). Although EndoMT was initially hypothesized to be active solely during development, recent studies suggest the contrary. Although described in various animal models of fibrosis across disciplines,187–189 controversy exists regarding EndoMT and its role in fibrotic disease. 190 In the context of renal pathology, EndoMT has been characterized in CKD as a source of fibroblasts and myofibroblasts. 191
EndoMT during fibrosis is stimulated through induction of TGF-β, which may originate from circulating inflammatory cells or mesenchymal cells. 192 Wang and colleagues recently investigated EndoMT in chronic allograft rejection and found the transition to be driven by TGF-β, Smad, and the Akt/mTOR/p70S6K signaling. 65 EndoMT has also been examined in models of CKD, UUO, diabetic nephropathy, and Alport syndrome, in which a portion of fibroblasts were found to have endothelial origin. Interestingly, these models produced various amounts of EndoMT-derived fibroblasts, suggesting specific stimuli may be responsible for EndoMT induction and the level of such induction. 191
Macrophage-To-Myofibroblast Transition (MMT)
MMT is a process by which macrophages differentiate into myofibroblasts (Fig. 3). In a recent study, cell lineage tracing demonstrated that bone marrow-derived macrophages undergo differentiation in to myofibroblasts during murine UUO. Interestingly, it was found that >60% of collagen-producing (α-SMA+) cells were derived from M2 (alternatively activated; anti-inflammatory) macrophages. 193
Furthermore, Wang and colleagues examined human renal allograft biopsies and demonstrated that macrophages (CD68+) actively underwent transition into myofibroblasts (α-SMA+), similar to findings in murine UUO. Fate mapping showed that bone marrow-derived macrophages were able to differentiate into myofibroblasts, which was prevented by Smad3 deletion, 194 highlighting the potential importance of the contribution of MMT in the development of renal fibrosis.
In conclusion, inflammation is a formal recognition of damage to renal tissue and is a normal physiological process required to resolve injury. Inflammation is initiated by renal insult and involves highly regulated cytokine and chemokine release, which bridles the inflammatory response to carefully orchestrate the injury response via recruitment, activation, and then suppression of inflammatory cells. 195 Activation of key signaling pathways can either induce a cascade of adaptive or maladaptive repair mechanisms. Inflammation plays a key role in the development of renal fibrosis and CKD; however, the exact nature by which this occurs remains ambiguous. It is clear that not one single cell type, factor, or pathway can be manipulated to prevent renal fibrosis, and further, the complex dynamics of the milieu involved in responding to injury can have completely different impacts on progression, depending on the type and stage of disease.
In summary, a more in-depth understanding of how inflammatory and fibrotic pathways can be manipulated for therapeutic intervention in the setting of renal diseases is key for the advancement of this field. Importantly, these pathways are of biological relevance and allow for proper healing when controlled. Future work should acknowledge the double-edged sword of renal inflammation and fibrosis. Studies should focus on regulatory mechanisms to control temporally persistent activation of pro-inflammatory and pro-fibrotic pathways, understanding that inflammation is necessary for the injury response but that it must resolve in a timely manner to prevent maladaptive tissue fibrosis.
Footnotes
Competing Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: AA serves as a consultant for DynaMed and is on the advisory board of Goldilocks Therapeutics.
Author Contributions
LMB, JML, and AA wrote, edited, and approved the final version of this manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by VA Merit Award I01-BX004047 (to AA) and National Institutes of Health Grants R01-DK-59600 (to AA), U54-E530246 (to AA), and P30-DK079337 (to AA), T32-DK-116672 (to LMB), F31-DK-115169 (to JML), and T32-GM-008361 (to R Lorenz for JML).