
Abstract
Glanzmann's thrombasthenia (GT) is an inherited, intrinsic platelet function defect that involves the platelet glycoprotein complex IIb–IIIa, also known as the fibrinogen receptor and the integrin αIIbβ3. The defect was originally described by Dr. Glanzmann in humans in 1918 as a bleeding disorder that differed clinically from other known coagulopathies. Over the decades that followed, researchers determined the biochemical and molecular basis for the disease in humans. Otterhounds with thrombasthenic thrombopathia, described in the 1960s, were the only animal model that closely resembled the disease described in humans until 1996. At that time, a Great Pyrenees dog was identified with unequivocal clinical and biochemical features of Type I GT. The cDNA encoding for glycoproteins IIb and IIIa were sequenced in normal dogs in 1999, allowing for identification of specific mutations causing Type I GT in both Otterhounds and Great Pyrenees dogs. Knowing the molecular basis for Type I GT in dogs as well as the cDNA sequences in normal dogs should enhance the understanding of structure/function relationships of the αIIbβ3 integrin and provide an excellent animal model for studies aimed at correction of GT in humans. The following review focuses on the structure and function of this platelet receptor and reviews the molecular, biochemical, and clinical aspects of Glanzmann's thrombasthenia in humans and dogs.
Glanzmann's thrombasthenia (GT) is an inherited, intrinsic platelet function defect involving the glycoprotein complex IIb–IIIa, also known as the αIIbβ3 integrin. The defect was originally described by a Swiss physician, Edward Glanzmann, 60 in 1918; however, the biochemical basis for the defect was not understood until the mid-1970s. 98,102,105 The first genetic mutation causing GT in humans was described in 1990. 18 Bleeding was described as being primarily mucosal, with epistaxis being the most common clinical manifestation. Bleeding patterns were consistent with laboratory findings that revealed normal platelet numbers but lack of platelet aggregation in response to all agonists and severely impaired clot retraction. Coagulation screening tests and von Willebrand factor antigen levels were also normal, ruling out other causes for the bleeding disorder. Bleeding tendencies in affected individuals ranged from mild to severe, with males and females being equally affected, suggesting an autosomal inheritance pattern. Although early reports of the defect were almost entirely within consanguineous populations, today the defect has been described in numerous ethnic groups, many without known consanguineous relationships. 49 In veterinary medicine, Otterhounds were the first breed described with a platelet defect that closely resembled that described in human GT. 38 Subsequently, platelet defects were described in Basset hounds and Great Pyrenees dogs that suggested these defects were also possibly GT. 15,28 Molecular studies confirmed that thrombasthenic thrombopathia of Otterhounds and the platelet defect in Great Pyrenees were identical to Type I GT described in human beings (Table 1). 16 Molecular studies of the Basset hound defect, however, did not confirm the existence of GT.
Comparison of canine von Willebrand's disease (vWD) with canine and human Type I Glanzmann's thrombasthenia (GT) and human variant GT.∗

N = normal; A/R = absent/reduced; N/R = normal/reduced; R = reduced; LN = low normal; A/I = absent/increased; Y = yes.
The vitronectin receptor may be increased with defects in the gene encoding for IIb.
Studies of GT at the molecular and biochemical level have led to a better understanding of the structure–function relationships of integrins in general, but especially αIIbβ3. Expanded capabilities in terms of diagnostics, drug development, treatment, and disease prevention in areas ranging from venomology 110,112 to cardiology 106 have occurred as a result of such studies. Comparative information on this integrin across species lines should further enhance our knowledge, particularly at the molecular level, which will aid in prediction of amino acid arrangements and protein structure, phenotype, and immunologic cross-reactivities. In cardiovascular medicine, much of the focus in recent years in humans has been on inhibiting platelet reactivity postmyocardial infarction. Many promising agents have been developed that target the αIIbβ3 integrin specifically 35,106,118 based on information known at the biochemical and molecular level in human platelets about this integrin. Most of these agents were evaluated for their efficacy in animal models before they were used in humans, yet very little comparative data were available at the molecular level in dogs, the animal model usually used. Perhaps having more information at the molecular level in animal models will shed light on why some of these agents, although seemingly efficacious and safe in dogs, were not found to be so in humans. Recently, researchers have suggested that certain polymorphisms within the β3 subunit protein predispose human beings to heart disease. 22,131,133 The most controversial report by Weiss in 1996 identified the PlA2 allele as a significant risk factor for acute myocardial infarction. Since that time, numerous studies and reports have been presented, with approximately 50% of those reports supporting the original observation and another 50% refuting the observation. 22 Knowing phenotypes and genotypes in other species may help to clarify this and other areas of controversy. The sequences for the genes encoding for the αIIb and β3 subunits have been determined in mice. 31,126 β3 knockout mice have been developed that have clinical and laboratory features characteristic of GT, including mucosal hemorrhage, poor clot retraction, and reduced platelet aggregation. However, their use as a model for GT in humans is in question with the recent finding that these mice develop osteosclerosis, a syndrome not described in humans with GT resulting from β3 subunit defects. 91 αIIb knockout mice have also been recently developed by introducing the herpes virus thymidine kinase gene into the αIIb locus. 129 These mice have characteristic features of GT, including reduced alpha granular content of fibrinogen, further verifying the necessity of an intact αIIbβ3 integrin for the uptake and storage of fibrinogen. Although these mice may prove to be good models for GT, their size is a limiting factor, especially for experiments requiring repeated blood sampling. Recently, the cDNA sequences for the genes encoding the αIIb and β3 subunits were determined in the dog. In addition, the molecular basis for GT was determined in Great Pyrenees and Otterhounds. This information should help investigators continue to obtain a better understanding of the αIIbβ3 integrin, with continued progress in disease recognition, prevention, treatment, control, and potentially cure.
The αIIbβ3 Integrin
The αIIbβ3 glycoprotein, an extensively studied integrin, is associated primarily with megakaryocytes and their progeny, platelets. 106,121 αIIbβ3 is an abundant and functionally significant integrin expressed on platelets. 121 This receptor binds fibrinogen with highest affinity but also binds other Arginine-Glycine-Aspartic acid (RGD)-containing molecules such as fibronectin, von Willebrand factor, and vitronectin. 10,56,107,116,121 The αIIbβ3 integrin is required for platelet aggregation, 52 clot retraction, 34 and platelet spreading along vascular matrices 122 but also plays an important role in platelet transmembrane signaling 32,33,57,65,69,85,121 and in the uptake of fibrinogen for storage in platelet α-granules. 66 Platelets possess 40,000 to 80,000 molecules of αIIbβ3 per cell, 25,130 with most of these receptors being distributed on the membrane surface 71 and the remainder being bound to the inner membrane of α-granules 134 and the internal surface-connected canalicular system. 138 Upon platelet activation, the occult αIIbβ3 molecules are translocated to the platelet surface when the α-granules fuse with the platelet membrane and the canalicular system evaginates during the shape change response. 24,96 Platelet activation also initiates cytoplasmic signals that induce αIIbβ3 clustering and triggers conformational changes in αIIbβ3 extracellular domains (inside-out signaling) to enhance ligand binding. 24,123,124 The receptor clustering is mediated by cytoskeletal reorganization and structural changes in proteins that are directly or indirectly linked to the cytoplasmic tails of αIIbβ3. 47,84,121 Although precise details regarding αIIbβ3 and platelet inside-out signaling remain unclear, circumstantial evidence suggests that the mechanism is similar to that of other integrins. Platelets contain RhoA, FAK, and Src proteins that reportedly mediate integrin clustering, stress fiber formation, and focal adhesion assembly in other cells. 32,33,47,94,121 Phosphatidylinositol-kinase (PI-K) converts phosphatidylinositides to 4,5-bisphosphate (PIP2) and 3,4,5-triphosphate (PIP3). 75,121 Exposure of platelets to thrombin stimulates PI-K activation and results in αIIbβ3 activation, but PI-K inhibitors, when added to thrombin-treated platelets, decrease the ligand-binding affinity of αIIbβ3 and promote platelet disaggregation. 81,121,140
αIIbβ3-ligand association triggers receptor clustering on the platelet membrane, and the simultaneous binding of dimeric fibrinogen molecules to these receptor clusters mediates platelet aggregation. 24 Receptor-ligand binding also stimulates interactions between the αIIbβ3 cytoplasmic domains and the platelet cytoskeleton (outside-in signaling) and initiates clot retraction. 24 Although details are unclear, fibrinogen-binding and agonist stimulation initiate phosphorylation (activation) of Src, Syk, and FA kinases that activate other proteins and lead to further cytoskeleton reorganization. 24,121
The αIIbβ3 receptor was one of the first integrins identified, 98 purified, 73 cloned, 90,113 and sequenced 45 and was also the first integrin to be expressed in recombinant form. 59,100 In 1974, Nurden and Caen identified an abnormal migration pattern when platelet-associated glycoproteins from three thrombasthenic patients were processed and electrophoresed and compared to patterns obtained from normal platelets. Two of these glycoproteins, which were undetectable on platelets from the thrombasthenic patients, were eventually designated GPIIb and GPIIIa. 98 In the early 1980s, Jennings and Phillips purified glycoproteins IIb and IIIa from platelets and characterized the GPIIb-IIIa complex as being calcium dependent. 73
Expression of αIIbβ3 on the platelet surface requires the presence of both subunits; defects in either αIIb or β3 usually result in absence of the αIIbβ3 complex. 100 Maintenance of αIIbβ3 stability is calcium dependent and fibrinogen binding is optimized when all four αIIb calcium-binding sites are occupied. 63,135 When the αIIbβ3 complex is expressed on the platelet surface, the amino termini of the α and β subunits interact with each other to form a globular structure that contains the ligand-binding domain. 132
The αIIb subunit is synthesized as a single precursor protein that forms a dimeric complex with the β3 subunit while associated with the rough endoplasmic reticulum (ER). 100 The αIIbβ3 complex is transported to the golgi apparatus where the αIIb subunit is cleaved into a light chain of 137 amino acids and a heavy chain of 871 amino acids that remain linked by a disulfide bond. 24,44,78,79,114 The mature αIIb subunit possesses seven functional domains: extracellular, 24,78,79 transmembrane, 24,48 cytoplasmic, 24,48,64 and four extracellular calcium-binding domains. 24,63,68,108 The light chain makes up the αIIb cytoplasmic and transmembrane domains and a small portion of the extracellular domain. 77,97 The majority of the αIIb extracellular domain, including the four calcium-binding domains, is associated with the heavy chain 77,97 (Fig. 1).
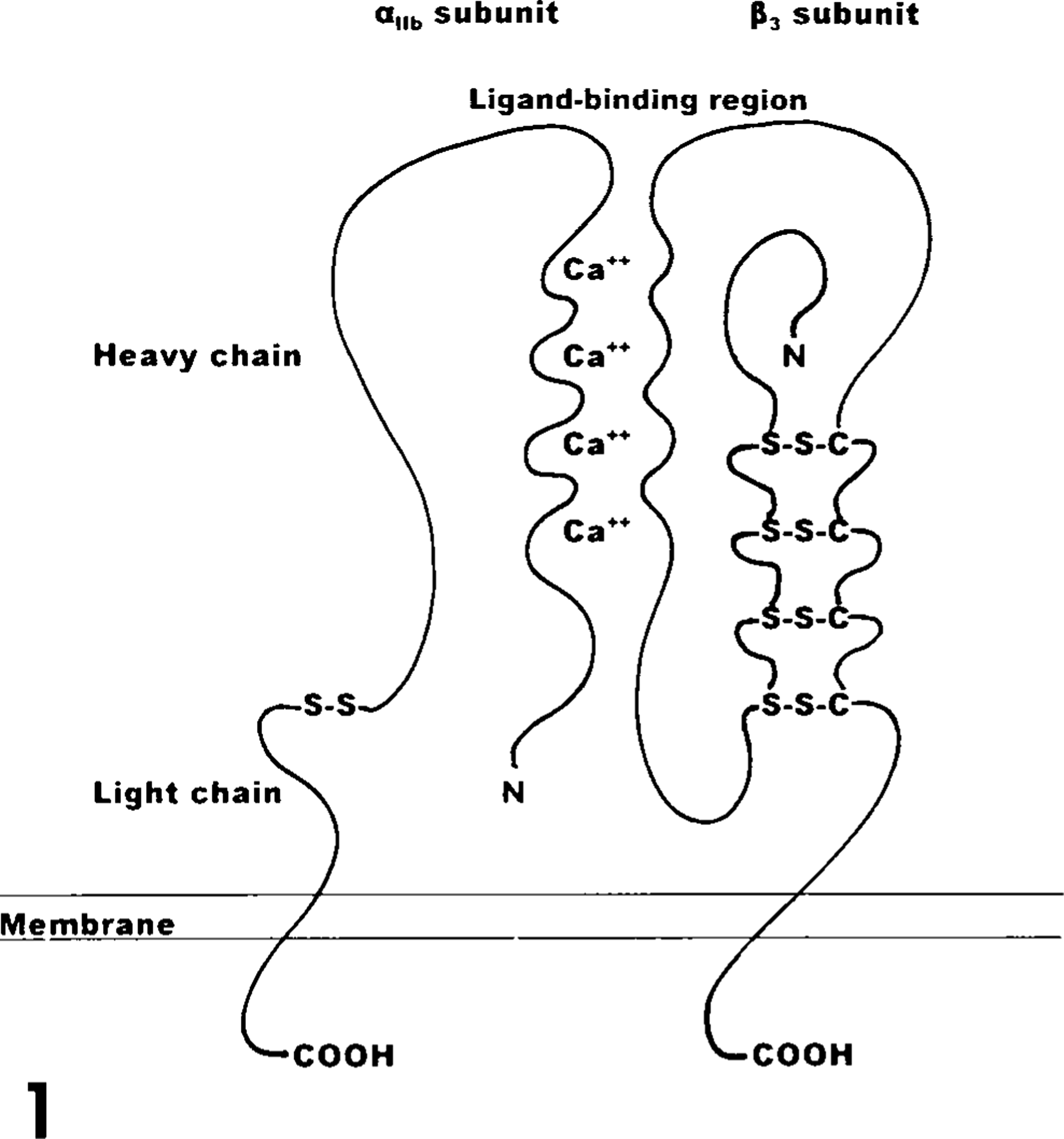
Structure of αIIbβ3 complex. The αIIb light chain, heavy chain, and four calcium-binding domains (Ca++) are designated. The amino termini are indicated by N and the carboxy termini are labeled COOH. Disufide bonds are indicated by (-S-S-). Diagram is modified from Albelda et al. 1 and Ginsberg et al. 59
The genes that encode the αIIb and β3 subunits have been mapped to the q21–23 band of chromosome 17 in humans. 19,125 The αIIb gene, from genomic DNA, consists of approximately 17.2 kilobases (kb) and contains 30 exons, designated 1 through 30, that range in size from 45 basepairs (bp) to 249 bp. 68,108 Each of the four calcium-binding domains consists of 12 amino acids that are encoded by gene segments of two adjacent exons. 108 The first calcium-binding domain (closest to the amino terminus) is encoded by exons 8 and 9; the second calcium-binding domain is encoded by exons 11 and 12; the third is encoded by exons 12 and 13; and the fourth is encoded by exons 13 and 14 108 (Fig. 2). The site of posttranslational cleavage into a light chain and a heavy chain is encoded near the 3′ end of exon 26 108 (Fig. 2). The αIIb gene has 5′ and 3′ untranslated regions (UTR) that are encoded by exons 1 and 30, respectively, and exon 1 encodes a 31 amino acid signal peptide 68,108 (Fig. 2).
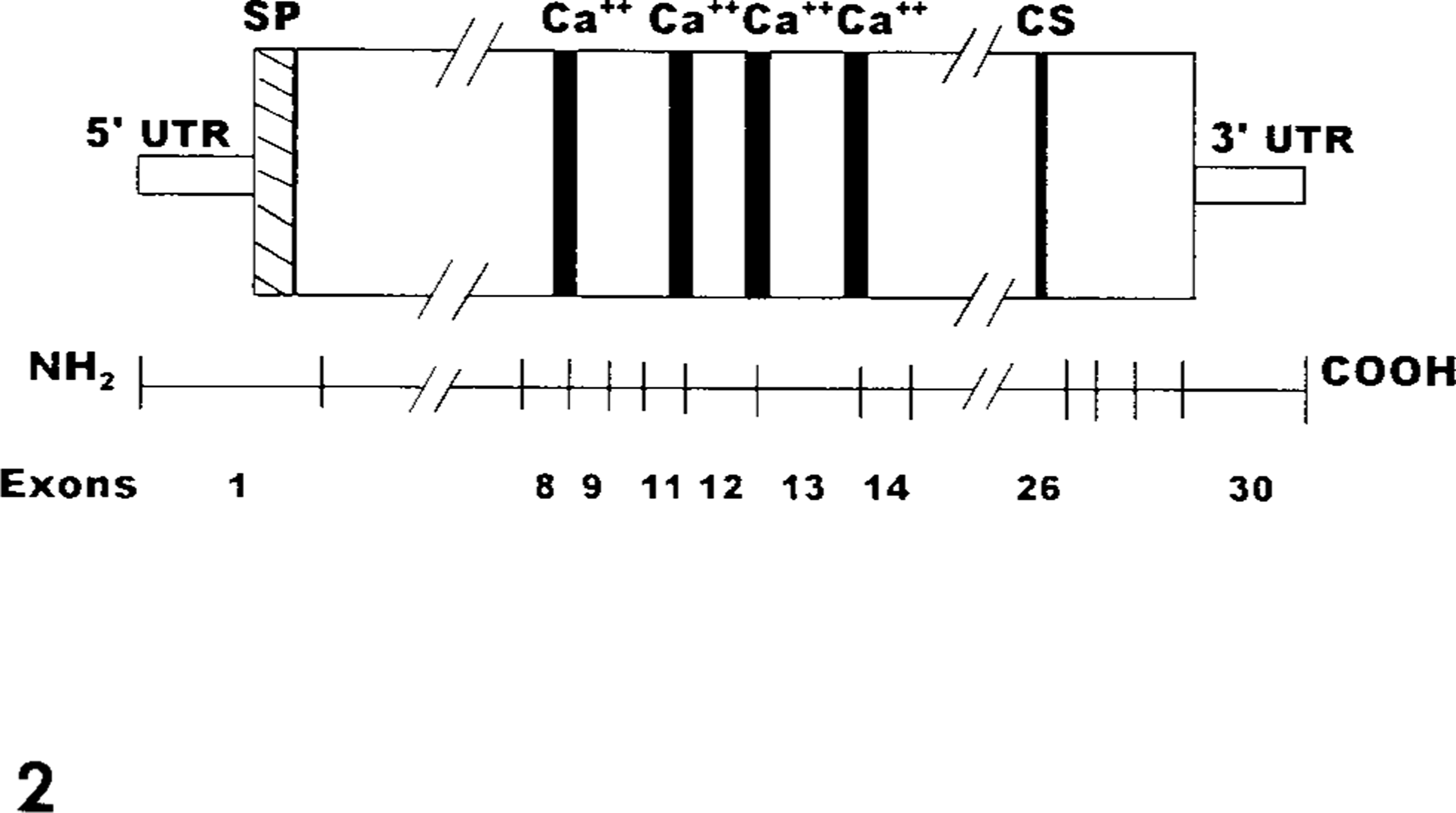
Relationship between αIIb coding region and exon distribution. Exon 1 encodes the 5′ untranslated region (UTR and the signal peptide (SP). The four calcium-binding domains (Ca++) are encoded by segments of exons 8, 9, 11, 12, 13, and 14. The cleavage site (CS) is encoded near the 3′ end of exon 26. Exon 30 encodes a 3′ UTR. Diagram has been condensed as indicated by (//) and is modified from Heidenreich et al. 68
The β3 subunit is a single polypeptide 762 amino acids in length 45 and has five functional domains: cytoplasmic, 139 transmembrane, 45 extracellular, 48 and RGD (ligand-binding) 39,41 domains and a region associated with calcium-dependent stabilization of the αIIbβ3 fibrinogen-binding pocket. 80,83,120 In addition, the β3 subunit possesses five extracellular cysteine-rich regions that facilitate disulfide bond formation and confer a globular conformation to the subunit 26,97 (Fig. 1). The β3 gene of humans is 63 kb in length and is composed of 14 exons, designated A through N, and range in length from 87 bp to 430 bp. 82,137,141 The β3 sequence, obtained from cDNA, is 2.3 kb in length. 31 Exons I and J encode four cysteine-rich regions that form loop structures via disulfide linkage with segments encoded by exons C, D, and E 83 (Figs. 1, 3). Exon A encodes a 26 amino acid signal peptide. 45 Protein segments important for ligand binding, the RGD-binding region and the segment associated with stabilization of the fibrinogen-binding pocket, are encoded by exons C and D, respectively 29,39,41,80,83 (Fig. 3).
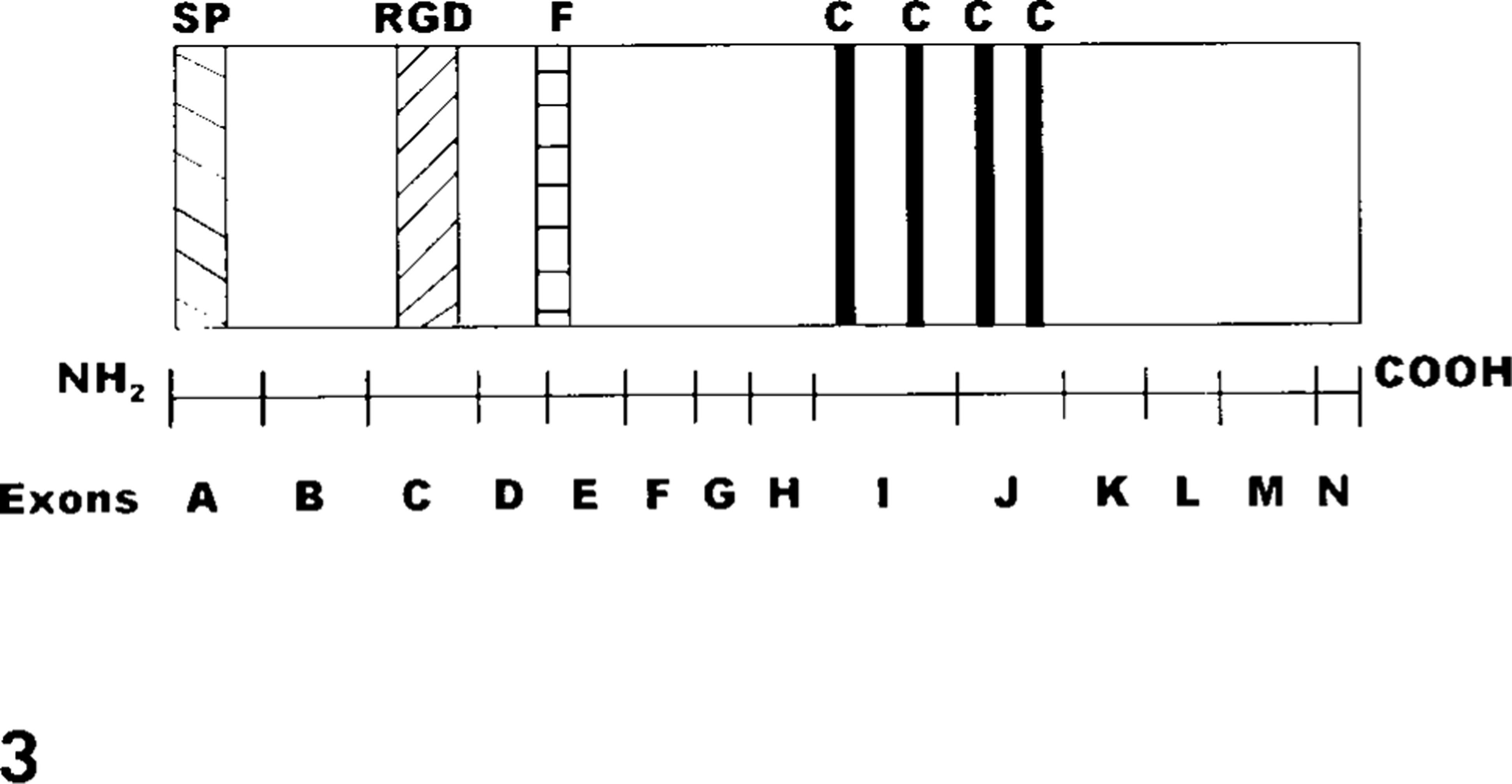
Relationship betweend the β3 coding region and exon distribution. Exon A encodes the signal peptide (SP). Four cysteine-rich regions (C) are encoded by exons I and J. The RGD-binding region (RGD) is encoded by a segment of exon C and a segment associated with stabilization of the fibrinogen binding pocket (F) is encoded by exon D. Diagram is modified from Lanza et al. 82 and is not drawn to scale.
The nucleotide sequence of canine platelet-derived cDNA was compared with the sequence previously established for αIIb of humans. 88 Canine αIIb, from platelet-derived cDNA, was nine nucleotides shorter in length than the human gene due to the lack of nucleotides corresponding to positions 2401 through 2403 (exon 24 in humans) in canine cDNA and because the termination codon in canine αIIb was located six nucleotides 5′ to that of humans (exon 30). The nucleotides flanking the missing codon in exon 24 are conserved in dogs and humans, and the negatively charged character of the carboxy-terminus is maintained by the deduced sequence of six consecutive glutamic acid residues in canine αIIb. The nucleotide sequence of full-length αIIb obtained from canine platelet-derived cDNA shared 86% similarity with that of humans. The nucleotide sequences of αIIb segments that encode functional domains in humans, including the four calcium-binding domains, shared ≥78% similarity when the canine sequence was compared with that of humans. The deduced amino acid sequence of canine αIIb shared 100% similarity with the segment that encodes the second calcium-binding domain of αIIb in humans.
The nucleotide sequence of canine β3 from platelet-derived cDNA was compared with that of humans and mice. 86 The sequence of full-length canine β3 shared 92 and 87% similarity with the sequences previously reported for humans and mice, respectively. The nucleotide sequence of canine β3 shared at least 85% similarity with the segments that encode functional domains in β3 of humans and mice, and the region associated with calcium-dependent stability was identical for all three species when deduced amino acid sequences were compared.
The observed sequence differences may contribute to species variations in receptor–ligand interactions and may partially explain differences in reactivity of platelets for differenct species. Gene sequence conservation between species implies structural and functional importance, and β3 regions of complete and near-complete similarity across species may represent β3-specific domains. 31
In recent years, analysis of natural and artificial mutations affecting αIIbβ3 have provided valuable information concerning integrin structure–function relationships. 24,51 Subunit synthesis and assembly of the αIIbβ3 complex have been studied extensively. Posttranslational cleavage of αIIb into light and heavy chains is required for expression of the αIIbβ3 complex on the platelet surface. 78,79,142 Maintenance of the structural integrity of αIIb calcium-binding sites dictates heterodimer conformation and expression on the platelet surface. 8,11,135 Structural stability and surface expression of αIIbβ3 requires the association of all three peptides, the αIIb light and heavy chains and β3. 27,48,78,79,100 Maintenance of αIIb and β3 disulfide bridges is required for proper folding and subunit stability. 30,95 The αIIbβ3 RGD-binding sites (amino acid residues 109–171 and 211–222 of β3) were defined when investigators performed chemical cross-linking studies and identified mutations that disrupted ligand binding and resulted in the bleeding disorder Glanzmann's thrombasthenia. 5,29,39–43,89,120
Glanzmann's Thrombasthenia—Humans
In 1918, Dr. Edward Glanzmann, a Swiss physician, used the (translated) terms “hereditary hemorrhagic thrombasthenia” to describe the bleeding diatheses of his patients. 54,60 Dr. Glanzmann attributed his patients' bleeding disorders to defective platelets because of abnormal in vitro clot retraction despite normal platelet quantitation. 54,60 Later, other laboratory manifestations were reported, including prolonged bleeding time and the observation that platelets appeared isolated rather than having pseudopodia and being clumped (aggregated) when blood smears of affected individuals were examined microscopically. 17,46,54,93 In the 1960s, investigators characterized Glanzmann's thrombasthenia (GT) as a platelet function defect resulting in abnormal aggregation due to an unidentified platelet membrane abnormality. 54,67,142 In the mid-1970s, Nurden and Caen, as well as Phillips et al., demonstrated that platelets from thrombasthenic patients possessed decreased amounts of glycoproteins IIb and IIIa. 98,103,105 When other scientists showed that platelets from thrombasthenic individuals were unable to bind fibrinogen, it was suggested that the platelet glycoprotein complex IIb–IIIa may serve as the fibrinogen receptor. 9,55 Thrombasthenia was proposed to have an autosomal-recessive mode of inheritance due to the following observations: males and females were affected in equal numbers, parents of thrombasthenic patients were asymptomatic, and 25% of the cases were associated with consanguinity. 23,54,67 Also, variability in the degree of clinical bleeding was reported; some patients presented with severe and potentially fatal hemorrhage while others demonstrated only mild bruising. 54
Glanzmann's thrombasthenia was first described at the molecular level in humans in 1990. 20 Currently, GT is widespread and has been recently reported in Japanese, 2,3,127 African American, 21 Indian, 76 mixed Caucasian, 8 Chinese, 61,62 French Gypsy, 117 Iranian, 101 and Algerian 92 populations. Although the overall incidence of GT is low, the reported frequency of GT approaches that of Von Willebrand's disease and hemophilia in consanguineous populations. 113 A website database has been established with mutation information on reported human cases of Glanzmann's thrombasthenia at http://med.mssm.edu/glanzmanndb.
The occurrence of hemorrhagic episodes during infancy and early childhood usually leads to diagnosis of GT before the age of 5 years; however, symptoms typically diminish as affected individuals approach adulthood. 53 Hemorrhage associated with GT occurs primarily in mucocutaneous tissues. 19 Observed bleeding patterns include epistaxis (most common), bruising, gingival hemorrhage, gastrointestinal hemorrhage, hematuria, menorrhagia, and hemarthrosis. 53 Spontaneous hemorrhage is uncommon in GT, but the most serious bleeding episodes occur after trauma or are exaggerated versions of normal physiologic bleeding; e.g., spontaneous shedding of deciduous teeth and minor surgical procedures, such as circumcision, commonly result in severe hemorrhage. 4,53,119 In affected women, menstruation and parturition represent particular risks for severe hemorrhage. 53
The severity of bleeding associated with GT is unpredictable, even when comparing thrombasthenic siblings of similar age. 53 Glanzmann's thrombasthenia is best managed with supportive medical care (blood transfusions and/or platelet transfusions) and by minimizing anticipated risks of hemorrhage with the use of platelet transfusions. 53 The most common complication is iron deficiency anemia secondary to chronic blood loss, and iron therapy (oral and injectable) is frequently required. 53 The development of platelet isoantibodies is associated with platelet transfusions. 53
The first classification scheme for GT, proposed by Caen, was based on platelet α-granule fibrinogen content and degree of clot retraction; however, current classification also includes αIIbβ3 quantitation. 19,23,98,103,104 To date, 59 different molecular defects resulting in GT have been identified in 48 kindred. 7,49 Nineteen of these cases have been compound heterozygotes (different mutation inherited from each parent) while 29 have been homozygous.
Type I GT represents the majority of reported cases and is characterized by severe deficiency (<5% of normal concentration) of immunologically detectable αIIbβ3, inability of activated platelets to bind fibrinogen, markedly reduced fibrinogen within platelet α-granules, and failure of platelets to aggregate or sustain clot retraction. 53,104 Platelets from patients with Type I GT due to a mutation in the gene encoding for αIIb often display increased levels of the vitronectin receptor. 37 This phenomenon is thought to be due to increased availability of β3 for binding to αv subunits. Detection of normal to increased β3 on the surface of Type I GT platelets lends support to the existence of an αIIb gene defect. 37 Gene defects that have resulted in Type I GT include deletions and insertions that caused alternative splicing, nonsense mutations that resulted in premature truncation of either αIIb or β3, as well as single nucleotide changes that resulted in single amino acid changes in areas critical for normal subunit stability and processing. 7,50,102 Mutations between and near the calcium-binding domain encoding regions of αIIb will result in Type I GT if the mutation results in an overall change in charge. 2,8,109,115,136 These mutations do not impair pro-αIIb synthesis or pro-αIIbβ3 complex assembly; however, the complexes are not transported from the ER to the Golgi.
Type II GT represents 14% of the reported cases. Platelets of individuals in this group possess 10 to 20% of the normal quantity of αIIbβ3 and show minimal fibrinogen binding, minimal aggregation, and abnormal clot retraction. 53,104 Type II GT cases most commonly arise when defects are present in the gene encoding for β3. 50 Of particular susceptibility are areas of the gene encoding for the RGD binding domain (amino acids 109–171). Within this domain is a MIDAS-like structure, DXSXS, involving amino acids 119 through 123. This domain is highly conserved among all beta subunits. Another critical domain is defined by the amino acids 211–222. Both of these sites are important for ligand recognition/binding and for cation binding. 128 Mutations in the RGD-binding domain region usually result in either Type II GT or variant GT. 58,72,99 One case of Type I GT was due to a mutation involving the change of a leucine to a tryptophan at position 117. 6 Site-directed mutagenesis studies indicated that this mutation resulted in malfolded αIIbβ3 heterodimers, which could not be transported to the platelet surface. Type II GT as a result of mutations in the gene encoding for αIIb have also been reported. 135 Mutations between and near the calcium-binding domain encoding regions that do not result in an overall change in charge result in Type II GT due to reduced and slower transport of the αIIbβ3 complexes to the surface. αIIbβ3 complex assembly and transport from the ER to the Golgi are not impaired with these defects. 115
Variant GT represents 8% of reported cases. 53,104 Platelets of these individuals possess 50 to 100% of the normal quantity of αIIbβ3 but demonstrate absent or minimal fibrinogen binding and aggregation, and results of clot retraction tests vary from normal to absent. 53,104 Whereas Type I and Type II GT are due to quantitative deficiency of αIIbβ3, variant GT is due to a qualitative defect of αIIbβ3. 53,104 As with Type II GT, variant GT is usually a result of a mutation in the gene encoding for β3. The CAM variant, 58 involving an amino acid change from aspartic acid to tyrosine at position 119 (within the highly conserved MIDAS-like motif) was one of the first cases of variant GT described at the molecular level. The Strasbourg variant, 83 a mutation resulting from a change in amino acid arginine to tryptophan at position 214, illustrated the importance of the RGD-binding region between amino acids 211 and 222. Aspartic acid 217 is thought to play a major role in cation coordination. Change to amino acid tryptophan would be predicted to destabilize the cation coordination mediated at this position. 128
Glanzmann's Thrombasthenia—Canine
Otterhounds
Thrombasthenic thrombopathia of Otterhounds was originally described in 1967. 38 Affected dogs exhibited mucosal bleeding and prolonged bleeding times that were aggravated by stress or surgery. The defect was described as being a combination of Bernard Soulier's disease (reduced/absent levels of platelet glycoprotein complexes Ib–IX) and Glanzmann's thrombasthenia. Thirty to 80% of platelets from affected dogs were described as being bizarre and giant, a characteristic typical of platelets from patients with Bernard Soulier's disease. Platelet glycoprotein studies, however, indicated that glycoproteins II and III were reduced while glycoprotein I was increased. 111 Platelet aggregation in response to all agonists was markedly reduced and clot retraction was minimal to absent. The defect was aggressively pursued and largely eliminated from the breed by the 1970s. However, in the late 1980s and early 1990s, descendents of originally described affected dogs were identifed with platelet dysfunction. Platelet aggregation responses to ADP, collagen, and thrombin were markedly reduced to absent, and intraplatelet fibrinogen and clot retraction were markedly reduced, as had been described in the original population. Platelet glycoprotein studies indicated that affected platelets had reduced to absent amounts of glycoprotein subunits IIb and IIIa, but changes in other glycoproteins were not seen. Flow cytometry studies did indicate the presence of glycoprotein IIIa on the surface of affected platelets, suggesting that the defect may involve the gene encoding for glycoprotein IIb. Platelet morphology and platelet size were normal, unlike what had been described in the original population.
Platelet-derived cDNA encoding for glycoproteins IIb and IIIa were sequenced in known normal, obligate carrier, and affected Otterhounds in 1999. A single nucleotide change G1193 → C (G1100 → C if leader sequence is not included) was detected in exon 12 of the gene encoding for glycoprotein IIb in the affected dog. The obligate carrier Otterhound was heterozygous for this change and the normal Otterhound was unchanged compared with normal dogs. 12,13,16 This nucleotide change would result in the substitution of a histidine for an aspartic acid at position 398 (367) within the third calcium binding domain of glycoprotein IIb. Based on studies in humans, such a change would be expected to destabilize the glycoprotein IIb–IIIa complex, resulting in lack of expression of the complex on the platelet surface.
Great Pyrenees
The test case, first described in 1996, presented with a history of gingival bleeding and chronic epistaxis since the age of 6 months. 15 Platelet numbers ranged from 150,000/µl to 250,000/µl and were not low enough to account for the protracted mucosal bleeding seen. Von Willebrand factor antigen levels were normal. Platelet aggregation responses to ADP, collagen, PAF, and thrombin were characterized by shape change with no aggregation. Clot retraction was absent. Platelet membrane surface labeling with I 125 did not demonstrate detectable levels of glycoproteins IIb or IIIa; however, other glycoproteins were present in normal amounts. Flow cytometry studies were able to detect small amounts of platelet glycoprotein IIIa, suggesting a defect in the gene encoding for IIb.
Segments of platelet-derived cDNA from the affected dog encoding for glycoproteins IIb and IIIa were amplified and sequenced. 86,88 Affected dog cDNA contained a 14 base pair repeat in exon 13 and defective splicing of the intron between exons 13 and 14. The insertion disrupted the cDNA segment that encodes the fourth calcium-binding domain, caused a shift in the reading frame, and resulted in a premature termination codon after 42 aberrant codons. The truncation of the αIIb protein would be expected to completely eliminate the transmembrane and cytoplasmic domains and a large portion of the extracellular domain, including the fourth calcium-binding domain, which is important for intracellular processing of αIIbβ3 and transport of the complex to the platelet surface. Based on studies in humans, although assembly of the glycoprotein complex probably occurs, transport to the platelet surface is likely impaired.
Since this test case was described, a young male Great Pyrenees from the Chicago area was identified with a history of excessive bleeding during tooth eruption. 87 Platelet numbers and von Willebrand factor antigen levels were normal. The veterinarian was not in close proximity to a laboratory that could perform platelet function studies on the dog. Genetic analysis revealed a defect identical to that described in the test case. Gene studies on the parents and siblings revealed that the parents were carriers. Three siblings were also carriers and one was normal. One puppy died at birth and could not be tested. Great Pyrenees dogs related and unrelated to this family group have since been identified as carriers for this defect in Oklahoma, Indiana, and Illinois.
Basset Hounds
Basset Hound thrombopathia was first described in 1979. 74 Affected Basset Hounds experienced epistaxis, gingival bleeding, and petechiation on mucous membranes and skin. Platelet counts, von Willebrand factor analysis, and coagulation screening tests were normal while bleeding times were prolonged, tending to focus the cause of the bleeding disorder at the platelet function level. Platelet aggregation responses to most agonists was minimal; however, platelets did aggregate in response to thrombin with a characteristic lag phase. 28 Platelet α-granule fibrinogen content was determined to be normal as were membrane glycoproteins IIb and IIIa. Fibrinogen binding by platelets, however, was impaired. 28 These findings suggested that Basset Hound thrombopathia may be a form of variant Glanzmann's thrombasthenia. However, sequence analysis of platelet-derived cDNA collected from affected Basset Hounds was identical to normal dogs for the segments encoding for glycoproteins IIb and IIIa (Boudreaux, unpublished findings). Although Basset Hound thrombopathia appears to be a qualitative defect involving the glycoprotein IIb–IIIa complex, the defect cannot be placed in the variant GT classification if the same rules used in humans are applied to dogs. Studies have indicated that the failure of the glycoprotein IIb–IIIa complex to be expressed properly in response to most agonists may be due to a defect in the metabolism of cAMP. 14 Thus far, platelet defects in humans involving signal transduction-related causes have not been classified as variant GT.
Conclusions
Extensive study of the pathophysiologic mechanisms of GT have led to important discoveries regarding normal platelet function; e.g., αIIbβ3 was identified as the platelet receptor for fibrinogen. 98 Subsequent investigation of αIIbβ3 helped define the integrin family of adhesion molecules and also helped characterize receptor-ligand structure–function relationships. 70 Recent molecular-level studies of GT have increased our knowledge of the genes that encode αIIbβ3 and have also led to the identification of specific genetic mutations that are responsible for the thrombasthenic phenotype. 50
Determination of the nucleotide sequence of αIIb and β3 for different species may provide valuable information regarding αIIbβ3 structure–function relationships and should promote comparative studies that will increase the understanding of variation in integrin-ligand interactions. Knowledge of αIIb organization for different species may also improve the understanding of regulation of tissue-specific gene expression. Comparative analysis of αIIb and β3 for different species may be useful in studies of bleeding disorders that affect humans and veterinary species. Information gained may help establish the canine as a good model for gene therapy of Glanzmann's thrombasthenia and for evaluating αIIbβ3-inhibitors as antithrombotic agents.