
Abstract
Persistent hyperplastic tunica vasculosa lentis and persistent hyperplastic primary vitreous are congenital ocular anomalies that can lead to cataract formation. A line of insertional mutant mice, TgN3261Rpw, generated at the Oak Ridge National Laboratory in a large-scale insertional mutagenesis program was found to have a low incidence (8/243; 3.29%) of multiple developmental ocular abnormalities. The ocular abnormalities include persistent hyperplastic primary vitreous, persistent hyperplastic tunica vasculosa lentis, failure of cleavage of the anterior segment, retrolental fibrovascular membrane, posterior polar cataract, and detached retina. This transgenic mouse line provides an ontogenetic model because of the high degree of similarity of this entity in humans, dogs, and mice.
Keywords
Persistent hyperplastic tunica vasculosa lentis and persistent hyperplastic primary vitreous (PHTVL/PHPV) are congenital ocular anomalies that occur in dogs, 1 29 36 humans, 22 35 and a line of transgenic mice. 11 34 37 Sequelae resulting from this anomaly include posterior subcapsular cataract, retrolental fibrovascular membrane causing leukocoria, and secondary retinal detachment. 8 23 29 36 This entity has been shown to have a hereditary basis in two breeds of dogs, the Doberman Pinscher and the Staffordshire Bull Terrier, 10 19 33 but a specific genetic basis has not been found. The morphogenesis of PHTVL/PHPV has been described previously in dogs with the use of transmission and scanning electron microscopy. 5 6 31
TgN3261Rpw is a line of insertional mutant mice generated at the Oak Ridge National Laboratory. The TYBS transgene was used to generate these mice by pronuclear injection, 24 resulting in pigmented transgenic mice and white wild-type mice. We discovered that 3.29% of the transgenic mice were runted with multiple ocular anomalies. We characterized their ocular anomalies and found persistent hyperplastic primary vitreous, persistent hyperplastic tunica vasculosa lentis, failure of cleavage of the anterior segment, retrolental fibrovascular membrane, posterior polar cataract, and detached retina.
Materials and Methods
One hundred TgN3261Rpw mice and wild-type littermates were examined by slit lamp biomicroscopy as described below, and 300 TgN3261Rpw mice and wild-type littermates were examined histologically over a 3-year period. Of these 300, 57 were wild type and the remaining 243 were transgenic mice. Mice were genotyped by Southern blot analysis. Briefly, high molecular weight DNA was prepared by digestion of 1-cm tail segments by a standard protocol 25 and subjected to restriction enzyme digestion, agarose gel electrophoresis, transfer to nylon membrane, and hybridization to a 32P-labeled TYBS probe by standard techniques. 30
We examined 10 wild-type and 90 transgenic mice by slit lamp biomicroscopy prior to euthanasia, and eight transgenic mice (15 globes) were determined to have the clinical and histologic ocular manifestations of persistent tunica vasculosa lentis. Clinical examination was performed with one drop of 1% tropicamide (Tropicamide 1%: Bausch & Lomb®, Tampa, FL) instilled in each eye 30 minutes before examination. A direct ophthalmoscope was used to assess the presence of a fundic reflex. The mice were then examined with a Top-Con slit lamp biomicroscope (Top-Con, Tokyo Optical Co. Ltd, Tokyo, Japan). Structures evaluated were the cornea, depth of the anterior chamber, amount of iris dilation, and the lens. Mice were also assessed for behavioral differences.
None of the wild-type mice exhibited any abnormality. Mice were euthanatized either when they exhibited signs of disease (dyspnea, abdominal distension, or cachexia) or when they reached approximately 13 months of age. Method of euthanasia was cervical dislocation.
Eyes from 300 wild-type and transgenic mice were enucleated and placed in 10% buffered neutral formalin. After fixation, eyes were embedded in paraffin, sectioned, stained with hematoxylin and eosin, and examined by light microscopy. Abnormal eyes were also stained with Jones–Methenamine silver stain to accentuate the basement membranes.
Ten-, 16-, and 21-day-old unaffected transgenic mice and wild-type mice (three mice per group) and one 3-month-old affected transgenic mouse were euthanatized, and the eyes were enucleated and immediately placed in phosphate-buffered saline solution (PBS, pH 7.2). A puncture was made adjacent to the optic nerve with a 27-G needle. Fine dissecting scissors were used to cut from the needle hole to the equator. The cut was then continued around the equator. Eyes were placed in hematoxylin (Biogenex, San Ramon, CA) for 5 minutes, rinsed in PBS, and examined under the dissecting microscope. After examination, eyes were fixed in 4% formalin for 30 minutes and stored in PBS.
All animals were cared for in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and in compliance with federal, state, and local regulations.
Results
Southern blot analysis confirmed that the pigmented mice carried the transgene, TYBS and the wild-type mice did not. Clinical examination found that 8/90 (8.89%) transgenic mice had a negative fundic reflection. These eight mice were also approximately 25% smaller than their normal littermates. The rest of the transgenic and wild-type mice (92/100; 92%) were positive for fundic reflection and were fully dilated 30 minutes after mydriasis when examined with a direct ophthalmoscope. A positive fundic reflection was seen as a red reflection of the choriocapillaris in response to focal light. Eyes from the eight affected transgenic mice either were partially dilated with dyscoria or could not be dilated at all and did not have a positive fundic reflection. One or both eyes showed evidence of cataracts. Younger (2–4 months) affected mice had immature posterior polar cataracts. Older (6–8 months) affected mice had mature or hypermature cataracts.
The eight affected transgenic mice behaved differently from the unaffected transgenic and wild-type littermates. The affected mice walked with one side against a cage wall and were disoriented when placed in an abnormal environment (outside their cages).
The 8/243 affected transgenic mice examined histologically showed numerous abnormalities. These abnormalities included PHTVL/PHPV that was usually bilateral (6/7 mice with both eyes examined), with cataracts (12/14 lenses), lenticonus posterior (3/14 lenses), lens rupture (2/14 lenses), posterior migration of lens epithelium (14/14 lenses), duplicated lens bows (5/14 lenses), microphthalmia (2/15 globes), partial retinal detachment (5/15), and severe retinal atrophy (12/15 globes).
PHTVL/PHPV was characterized by moderate to marked posterior retrolental fibrovascular connective tissue closely adherent and expanded peripherally to the lens equator (Fig. 1). The tunic was heavily pigmented in some cases (5/14; 36%; Fig. 2), and in one eyeball, there were large capillaries/small arterioles embedded in the central portion of the connective tissue, reminiscent of the embryologic remnant of the vasa hyaloidea propria. Posterior lenticonus (Fig. 1) was seen in three lenses, two from the same mouse; all had PHTVL/PHPV, and two of 14 had a ruptured lens capsule (Fig. 3). The retrolental fibrovascular membrane was also nonpigmented in some cases (Fig. 4). Microphthalmia was seen in two cases and was associated with severe fibrosis and collapse of intraocular chambers due to failure of cleavage of the anterior segment. There was severe retinal atrophy in one case. In all 14 lenses examined, there was lens epithelium at the posterior lens and, in some cases, thickening of the posterior capsule and duplication of the lens bow (5/14; 36%; Fig. 5). Cataracts ranged from mild Morgagnian globules and bladder cells to rupture and fibrosis in others. There was moderate to marked retinal degeneration due primarily to loss of photoreceptors and outer nuclear layer in all 15 eyeballs. Hereditary retinal atrophy is present in the founder FVB/N strain and was seen in transgenic mice without PHPV/PHTVL. In two cases, one with microphthalmia, there was severe retinal atrophy, suggesting that atrophy was exacerbated by PHPV/PHTVL.
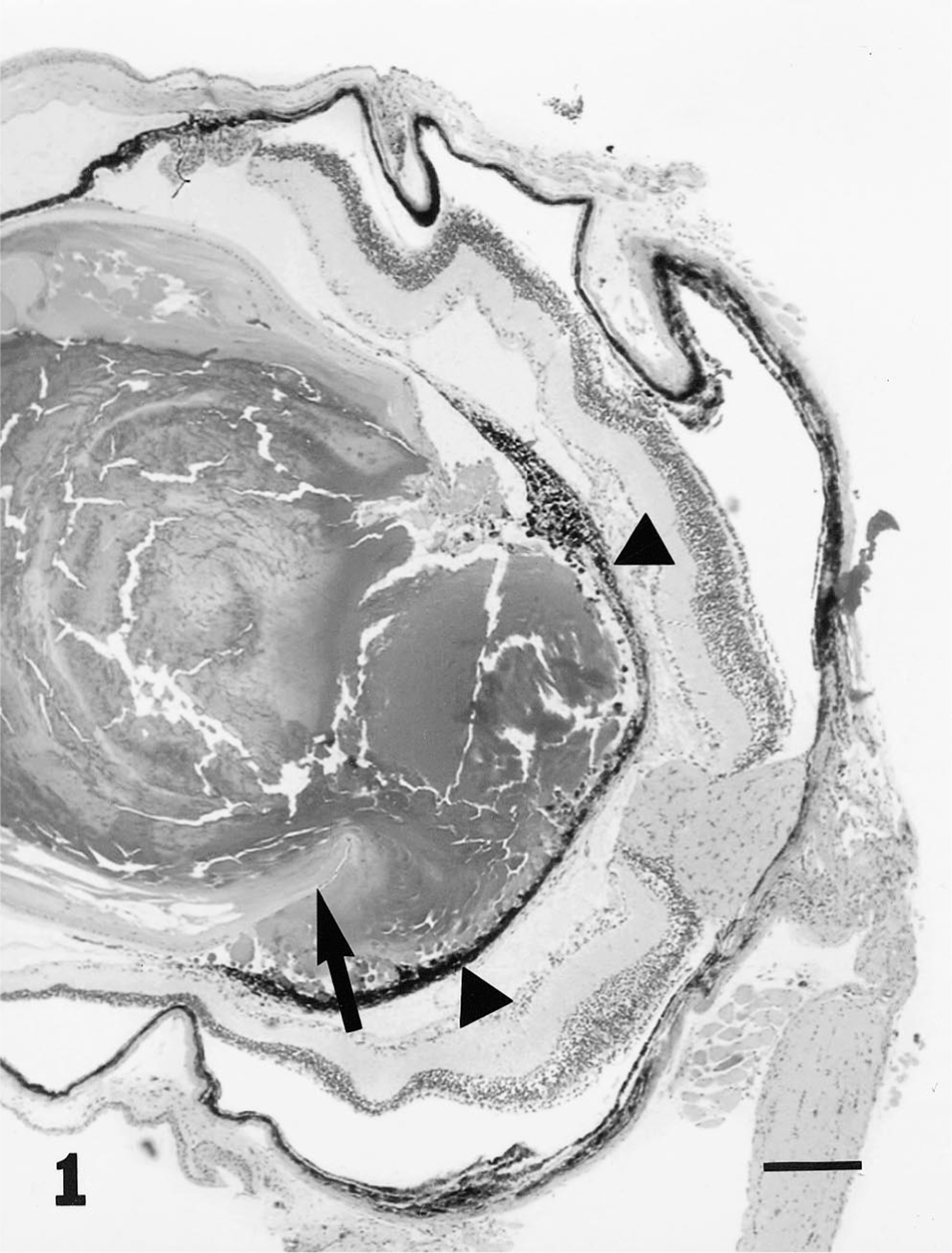
Eye; mouse. Photomicrograph of a globe depicting posterior lenticonus with lens capsule rupture (arrow) and posterior retrolental pigmented fibrovascular tissue (arrow heads). Bar = 380 μm.
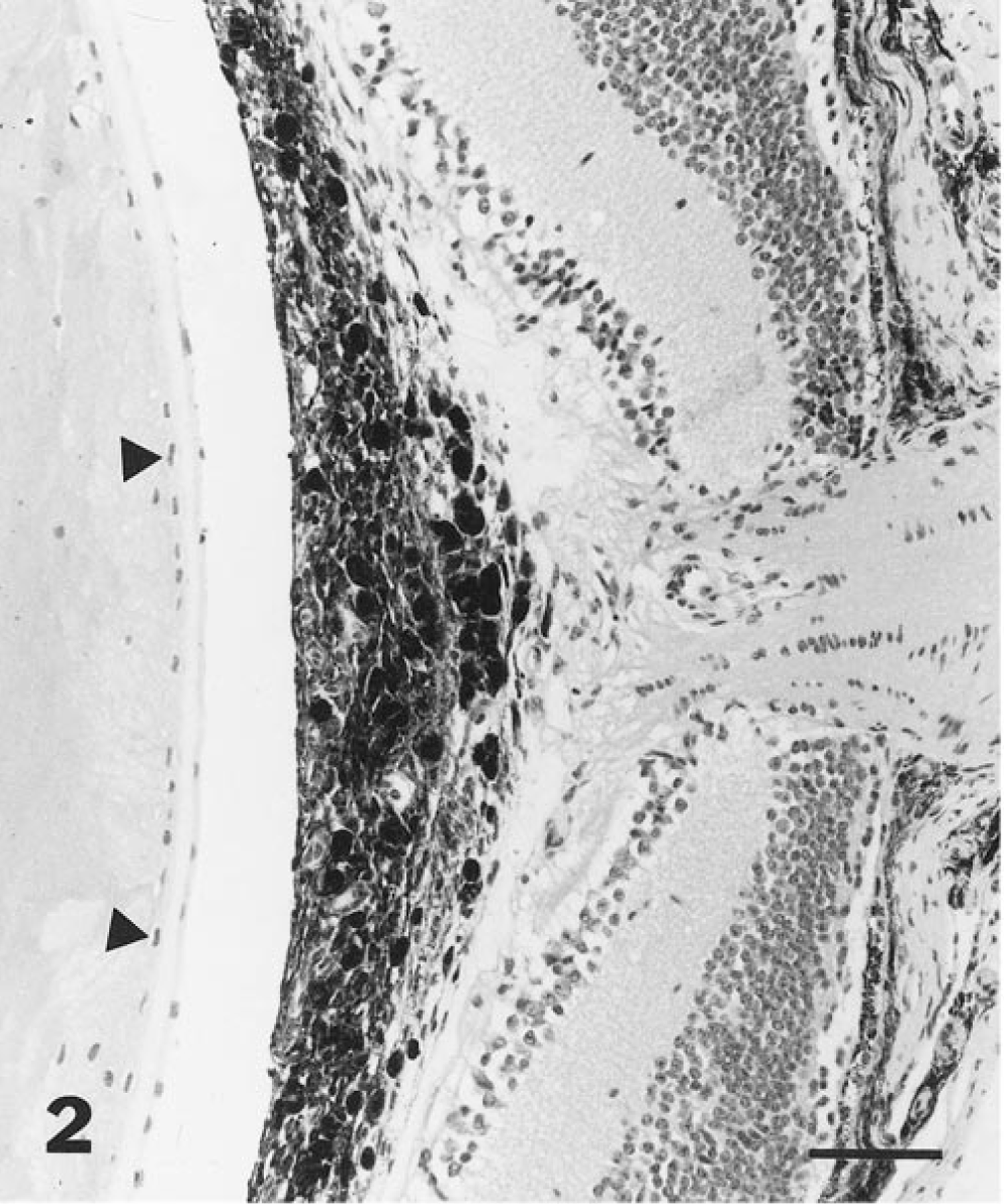
Eye; mouse. Photomicrograph depicting preretinal posterior retrolental pigmented fibrovascular tissue at the region of the optic nerve and posterior migration of lens epithelium (arrowheads). Bar = 60 μm.
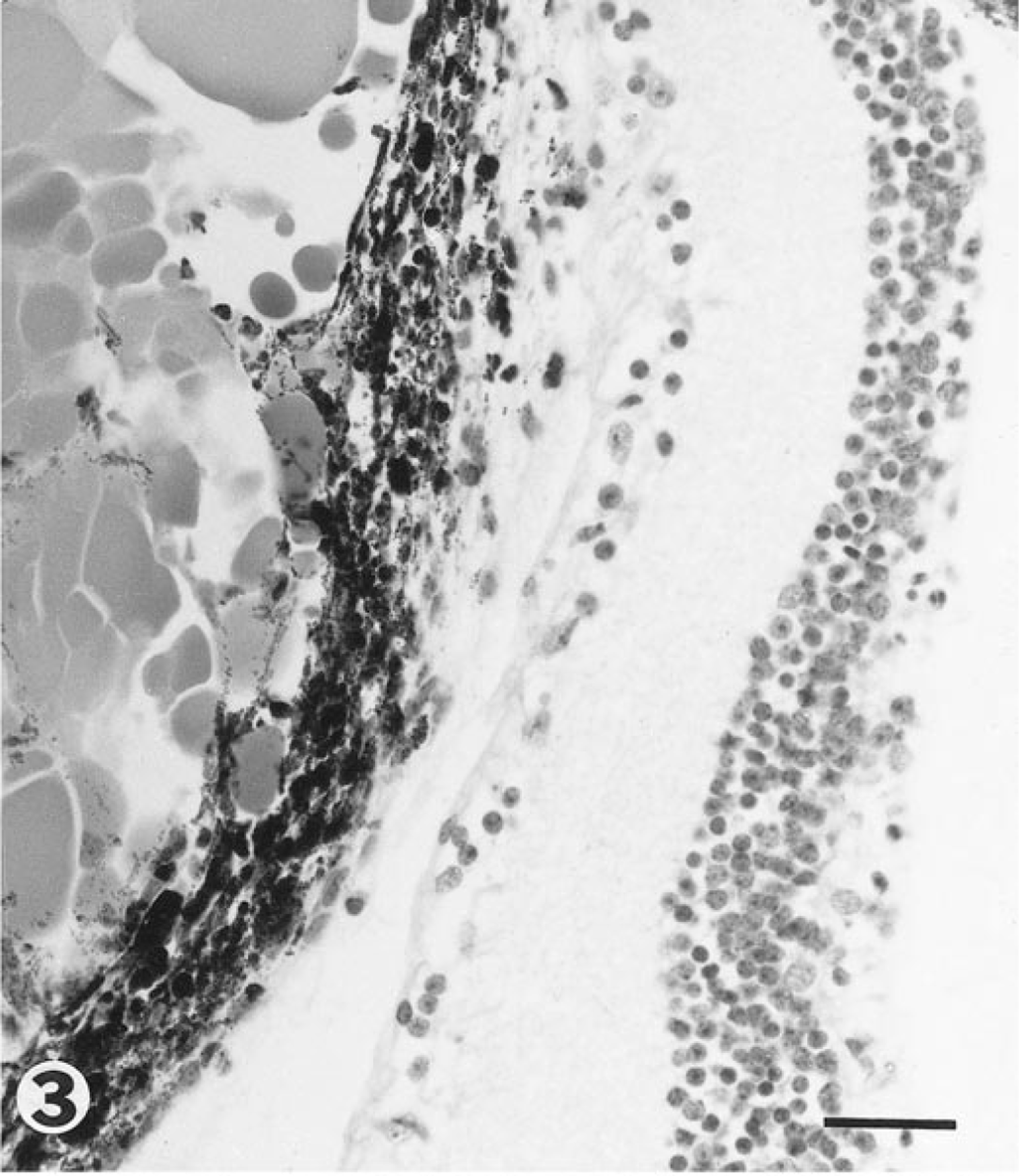
Eye; mouse. Photomicrograph of preretinal posterior retrolental pigmented fibrovascular tissue and globules of lens material released from ruptured lens. Bar = 30 μm.
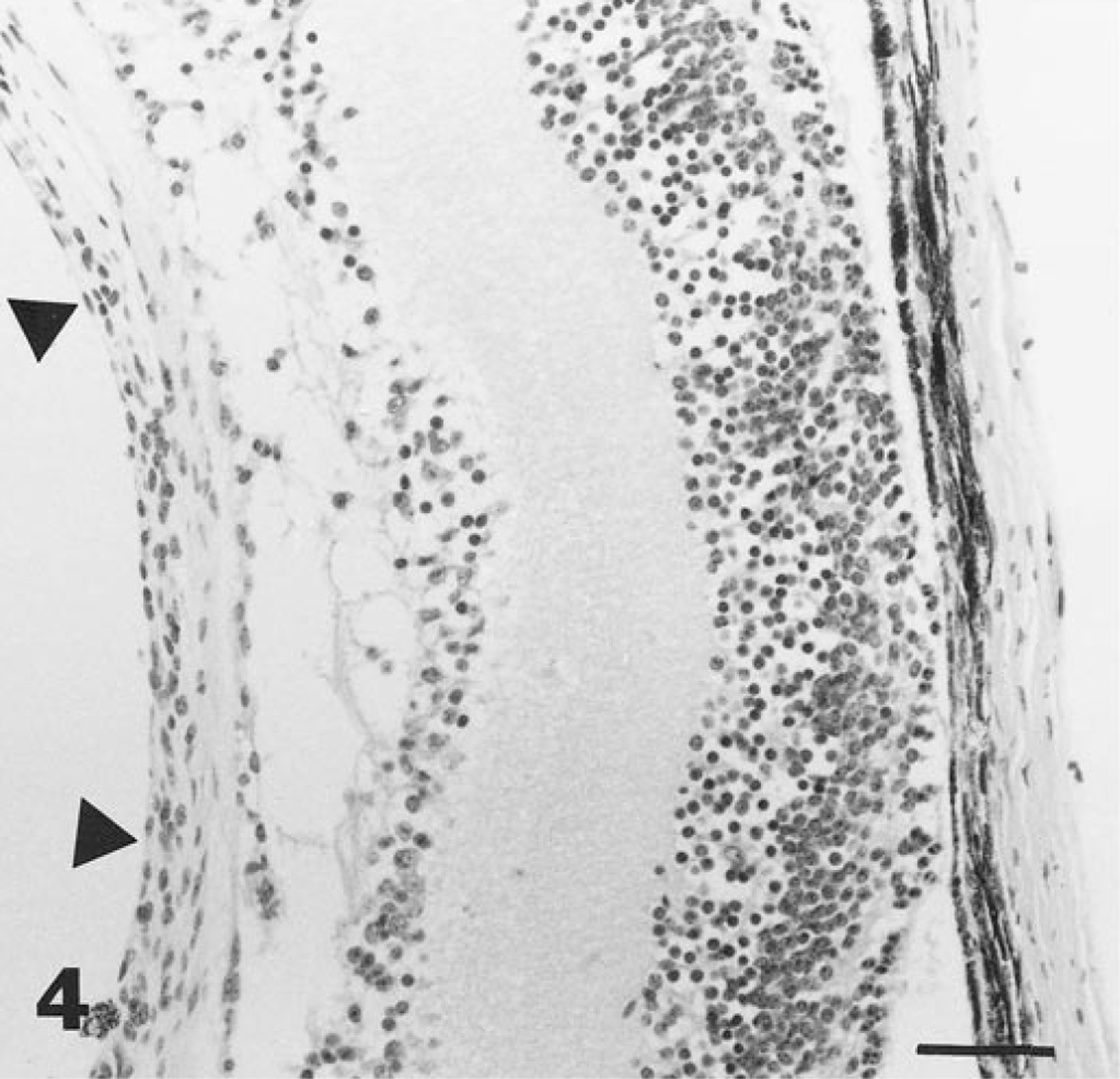
Eye; mouse. Photomicrograph depicting preretinal posterior retrolental nonpigmented fibrovascular tissue (arrowheads). Bar = 35 μm.
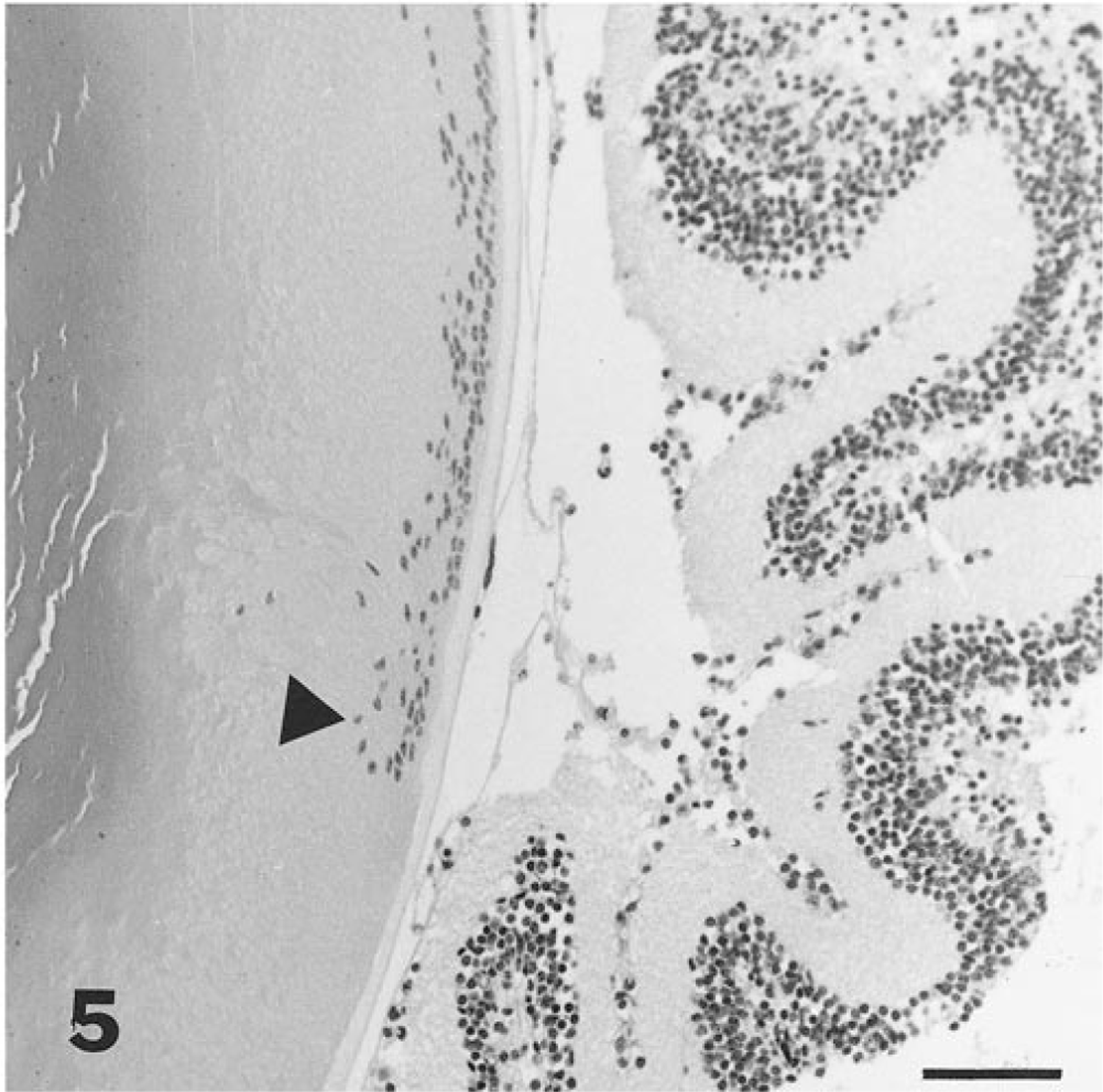
Eye; mouse. Photomicrograph of lens and retina with posterior migration and duplication of the lens epithelium (arrowhead). Bar = 55 μm.
The tunica vasculosa lentis in 10-day-old heterozygous and wild-type mice had already regressed. The primary vitreous was still present, with vessels coursing over the posterior lens capsule. The primary vitreous in 16-day-old heterozygous and wild-type mice was still evident; but in 21-day-old mice, the primary vitreous vasculature was mostly absent except for a few nonpatent strands.
The affected transgenic mouse examined did not have secondary vitreous present nor a space for it. The primary vitreous was thickened and attached to the retina and posterior lens capsule. A posterior polar cataract was evident in association with the primary vitreous. Examination of the anterior segment of the eye revealed a shallow anterior chamber and complete posterior synechia evidenced by the total attachment of the iris to the anterior lens surface.
Discussion
Ocular abnormalities found in affected TgN3261Rpw mice were similar to those described in dogs and in humans with PHTVL/PHPV. Our findings are similar to those reported in Doberman Pinscher dogs with PHTVL and the anterior form of PHTVL/PHPV in humans. 3 12 26–28, 31 This transgenic mouse line provides an ontogenetic model because of the high degree of similarity of this entity in humans, dogs, and mice.
Though grossly, histologically, and electron microscopically described in the literature, the actual cause has not been determined. 3 12 26–28, 31 PHTVL/PHPV has been postulated to be a result of faulty lens development. 3 31 Others have said that there is an increased growth promotion or decreased inhibition of intravitreal angiogenesis by growth factors. 4 20 21 Another longer believed hypothesis is that macrophages fail to induce regression of the hyaloid vascular system and its associated tunica vasculosa lentis by occluding the capillary lumens. 9 13 17 Others believed that the hyalocytes became phagocytically active in order to remove the hyaloid vasculature. 2 Lang and Bishop 15 generated a line of transgenic mice with a disrupted subset of macrophages that resulted in the persistence of the hyaloid vasculature and the pupillary membrane. More recently, it was shown that regression of the pupillary membrane occurs because of apoptosis induced by macrophages associated with the pupillary membrane. 16 This information, combined with that of Lang and Bishop, 32 suggests that macrophages play a key role in driving all aspects of pupillary membrane regression and possibly regression of the rest of the hyaloid vasculature.
In humans, PHTVL/PHPV is usually a unilateral nonhereditary condition. 28 In only 11% of cases, the condition is bilateral and is associated with severe malformations as in trisomy 13/15. 14 28 In the Doberman Pinscher and the Staffordshire Bull Terrier, PHTVL/PHPV occurs as a bilateral hereditary condition without other malformations and may be due to inbreeding. 32 Conventional karyotyping studies have not found any chromosomal aberrations in the Doberman Pinscher. 32 It is possible that PHTVL/PHPV is occurring in the Doberman Pinscher because of a mutation that is not evident by karyotype studies.
TgN3621Rpw mice not only develop ocular abnormalities but they also have a high incidence of histiocytic sarcomas (40%, unpublished data). These histiocytic tumors are of an undetermined lineage and occur in transgenic mice affected with ocular abnormalities and in nonaffected transgenics but not in wild-type mice. In light of the previously mentioned studies and the development of histiocytic-derived tumors, we propose that some TgN3261Rpw mice may have abnormally functioning hyalocytes as well as other tissue macrophages in the body. It is also possible that the insertion of the transgene is causing the abnormal function of the disrupted gene at a time that is crucial to ocular development. Perhaps a certain protein is present, absent, or mutated at a crucial moment that determines the first and most decisive event in development of ocular structures.
Transgenic heterozygous mice were used in the breeding program, and this should result in 25% of the offspring being homozygous, 50% being heterozygous, and 25% being wild type. We hypothesize that the runted mice affected with PHPV/PHTVL are homozygous. The extremely low incidence of the phenotype may be a result of either very low penetrance of the trait or loss of the mice at birth because of failure to thrive. None of the wild-type littermates was affected by any ocular abnormality except for retinal degeneration, further supporting that the ocular anomalies were due to the transgene.
This line of transgenic mice was generated on the FVB/N background, which has a mutation at the rd locus; that is, they have a form of retinal degeneration in which all rods and 97% of cones degenerate by 1 month of age, the same time at which normal murine retinas mature. 7 18 This mutation is common in many mouse strains and is due to a mutation in the β-subunit of the phosphodiesterase gene. 7 18 Though this theoretically makes the unaffected transgenic and wild-type mice blind by 28 days of age, they retain their pupillary light responses and behave normally when compared with mice without rd mutations.
In conclusion, Tg3261 is an interesting insertional mutant that requires further embryologic studies that will concentrate not only on the normal interaction of the hyalocytes with the other cells involved in normal ocular development but also on the abnormal migration of ocular cells occurring in the affected transgenic mice. Cloning the disrupted gene by molecular biology techniques will allow greater understanding of the ocular abnormalities and of the tumors.