
Abstract
Platelets are integral to normal haemostatic function and act to control vascular haemorrhage with the formation of a stable clot. The fibrinogen receptor (glycoprotein IIb/IIIa [GPIIb/IIIa]) is the most abundant platelet integrin and, by binding fibrinogen, facilitates irreversible binding of platelets to the exposed extracellular matrix and enables the cross-linking of adjacent platelets. The vital role of GPIIb/IIIa requires tight control of both its synthesis and function. After transcription from distinct domains on chromosome 17, the two subunits of the heterodimer are carefully directed through organelles with intricate regulatory steps designed to prevent the cellular expression of a dysfunctional receptor. Similarly, exquisite control of platelet activation via bidirectional signalling acts to limit the inappropriate and excessive formation of platelet-mediated thrombus. However, the enormous diversity of genetic mutations in the fibrinogen receptor has resulted in a number of allelic variants becoming established. The Pro33 polymorphism in GPIIIa is associated with increased cardiovascular risk due to a pathological persistence of outside-in signalling once fibrinogen has dissociated from the receptor. The polymorphism has also been associated with the phenomenon of aspirin resistance, although larger epidemiological studies are required to establish this conclusively. A failure of appropriate receptor function due to a diverse range of mutations in both structural and signalling domains, results in the bleeding diathesis Glanzmann's thrombasthaenia. GPIIb/IIIa inhibitors were the first rationally designed anti-platelet drugs and have proven to be a successful therapeutic option in high-risk primary coronary intervention. As our understanding of bidirectional signalling improves, more subtle and directed therapeutic strategies may be developed.
DECLARATIONS
None
The authors receive funding from Guy's and St Thomas’ Charity
N/A
CNF and AF
CNF performed the literature review. CNF and AF wrote the paper
Introduction
Platelets were first discovered over 130 years ago by Bizzozero,1,2 but it was not until the early 20th century that they were correctly identified as being derived from megakaryocytes, having been variously hypothesized as being fragments of leukocytes, extruded red cell nuclei and albuminous precipitants to name but a few. 3 Platelets are central to the formation of thrombus following vascular injury, 4 and have increasingly become a target for pharmaceuticals directed at cardiovascular disease prevention.
The platelet fibrinogen receptor is integral to the formation of platelet-mediated thrombus, as it represents the final common pathway of platelet activation, adhesion and aggregation. It is formed from two subunits of glycoprotein IIb (GPIIb; integrin ?IIb) and glycoprotein IIIa (GPIIIa; integrin ?3), and is the most abundant integrin on the platelet surface. Quantification of fibrinogen receptor expression using monoclonal antibodies has revealed that each platelet expresses approximately 80,000 such receptors on its surface, 5 with an additional internal pool that can be transported to the surface during platelet activation. 6
Despite the receptor's colloquial name, it is neither specific to platelets nor to fibrinogen, and the receptor is also found – unsurprisingly – on megakaryoctes 7 and also on some transformed cells. In addition to binding fibrinogen, 8 it also has the capacity to bind a number of other soluble and immobilized ligands, of which von Willebrand factor appears to have the most significant role in haemostasis. 9
This review highlights key points involved in the synthesis, function and regulation of the fibrinogen receptor in haemostasis, with reference also to the pathological fibrinogen receptor and to pharmacological attempts to manipulate its function.
Synthesis of the fibrinogen receptor
The fibrinogen receptor is formed from two calcium-dependent heterodimers10,11 that are encoded on the long arm of chromosome 17q21.32 with independent regulatory domains 12 (Figure 1). The synthesis of the two subunits differs, with GPIIb synthesized as a pro-GPIIb moiety composed of a heavy and a light chain connected by disulfide bonds,13,14 while GPIIIa is synthesized as a single, mature chain. 15
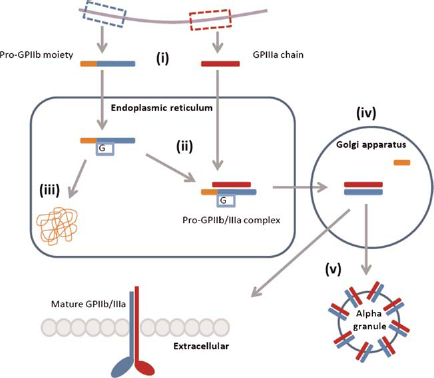
Synthesis of the fibrinogen receptor. (i) The subunits are transcribed from independent domains of chromosome 17q21.32, resulting in a pro-GPIIb moiety with heavy and light chains and a mature GPIIIa chain. (ii) The pro-GPIIb moiety is glycosylated and associates with the mature GPIIIa chain within the endoplasmic reticulum. (iii) Uncomplexed subunits are degraded by the calnexin cycle. (iv) Proteolytic cleavage of the pro-GPIIb subunit within the Golgi apparatus yields the mature GPIIb/IIIa. (v) Mature GPIIb/IIIa is transported to the plasma membrane or stored in alpha granules. G, glycoslation
Within the endoplasmic reticulum, the pro-GPIIb moiety associates with the mature GPIIIa after the latter has undergone post-translational glycosylation. 7 The formation of a pro-GPIIb/IIIa complex is a prerequisite for surface expression of either subunit, 16 as demonstrated in transfection experiments using COS cells. 17 Within the endoplasmic reticulum, high mannose N-linked oligosaccharides are added to the pro-GPIIb precursor 15 and uncomplexed sub-units are degraded in a proteasome-dependent manner controlled by the calnexin cycle. 18
The pro-GPIIb/IIIa complex is then transported to the Golgi apparatus where the mannose chains of the pro-GPIIb are converted into complex oligosaccharides, and proteolytic cleavage of the pro-GPIIb subunit yields the disulfide-linked light and heavy chains. This mature complex is then transported to the cell surface or stored within alpha granules.
The fibrinogen receptor in primary haemostasis
The central role of platelets is to terminate haemorrhage following vascular injury, 4 although recently significant roles in atherosclerosis, 19 immune response, 20 inflammation,21,22 tumour metastasis 23 and angiogenesis 24 have been identified. The involvement of the fibrinogen receptor in these diverse processes is currently poorly characterized although, as the primary mediator of platelet adhesion, and hence platelet localization, it is likely that the fibrinogen receptor participates to some extent. Indeed, the generation of integrin ?3-null mice in the investigation of Glanzmann's thrombasthaenia (GT) has revealed impaired neovascularization of the retina, 25 failure of the coronary capillaries to mature, 26 accelerated atherosclerosis, 27 cardiac hypertrophy and inflammation 28 and promotion of tumour growth 29 in such mice. Detailed discussion on these secondary roles of platelets is outside the scope of this review, but these have been comprehensively reviewed elsewhere.30–32
In the resting state, platelets circulate without interacting with the vessel wall or each other, but following vessel injury and exposure of the extracellular matrix, a plethora of different platelet receptors interact with each other in a coordinated manner to facilitate tethering, rolling and adhesion to the vessel wall and a cascade of intricate signalling processes; these result in the recruitment and aggregation of platelets to form a stable, haemostatic plug. The role of platelets in primary haemostasis has been comprehensively reviewed by Broos et al., 33 but we will briefly summarize the three main processes in which the fibrinogen receptor is involved: (i) adhesion to the vessel wall, (ii) platelet aggregation and (iii) bidirectional signalling.
Adhesion to the vessel wall
In order for platelets to adhere to the exposed extracellular matrix, they must be able to form bonds that are stronger than the rheological forces opposing their interaction. GPIIb/IIIa irreversibly binds immobilized fibrinogen 34 thus achieving stable adherence to the vessel wall. However, within arterial vessels there are high wall shear forces due to rapidly flowing blood, and the slow rate of bond formation by GPIIb/ IIIa is insufficient to facilitate stable adhesion. To resolve this problem, the glycoprotein Ib-V-IX complex on platelets slows their flow rate along the vessel wall by binding von Willebrand factor in a process that exhibits fast association and dis-association. 34 Once slowed, GPIIb/IIIa has sufficient time to form the irreversible bonds required.
Platelet aggregation
Once bound to the extracellular matrix, the now activated platelet must recruit and bind other platelets in order to form a haemostatic plug. Selective fusion of cytoplasmic granules with the plasma membrane results in release of a diverse complement of proteins, 35 many of which act as soluble platelet agonists in an autocrine and paracrine manner. The signalling steps that lead to activation of GPIIb/IIIa are summarized below, but they result in the receptor binding soluble fibrinogen that is required for the stable cross-linking of adjacent platelets.36,37
The mechanism of aggregation is also affected by shear. At elevated shear rates (1000– 10,000 s−1) platelets bind to each other initially via von Willebrand factor receptors, as with adhesion to the vessel wall. 33 At even higher shear rates (>10,000 s−1), platelet aggregation can occur in the absence of activation in the presence of soluble von Willebrand factor. 33 These aggregates however are unstable as, without activation of GPIIb/IIIa, irreversible platelet–platelet binding does not occur.
Bidirectional signalling
Some integrins are constitutively active but others require signalling events to transform them from a quiescent to an active state, thus enabling them to modulate their function in a temporal and spatial manner. GPIIb/IIIa is in the latter category, as tight regulation of its activation prevents uncontrolled platelet aggregation and inappropriate intravascular thrombus formation.
In response to a stimulus, structural rearrangement transforms the integrin from a closed, low-affinity, bent conformation to an open, high-affinity, extended conformation by a switchbladelike movement of the integrin legs 38 in a process that is regulated by both inside-out and outside-in signalling.
Inside-out signalling
Inside-out signalling refers to processes that act on the cytoplasmic domain of integrins (the cytoplasmic tails [CTs]) and result in changes in the extracellular domain, facilitating structural rearrangement of the ligand-binding site. In the context of affinity maturation of the GPIIb/IIIa receptor, platelet agonists (such as adenosine diphosphate [ADP] or thrombin) and adhesion receptors (such as the GPIb-V-IX receptor for von Willebrand factor) activate cytoplasmic proteins that interact with the CTs of GPIIb/IIIa.
The CTs of the alpha and beta subunits are intimately connected via hydrophobic and electrostatic bonds that require complete disruption and the subsequent unclasping of CTs for full activation of the receptor. 39 Unclasping of these cytoplasmic tails is facilitated by talin-H that is formed following activation of these cytoplasmic pathways and displaces the binding of the alpha-CT from the beta-CT. 40 Once unclasped the CTs, which have no intrinsic enzymatic activity of their own, offer previously hidden binding sites to over 20 cytoplasmic proteins that are involved in perpetuating platelet activation. 41
Outside-in signalling
Outside-in signalling, as the name suggests, is the converse of inside-out signalling and results in spreading of the platelet. Ligand interaction via the extracellular domain results in changes within the CTs. Ligand binding may follow previous activation of the receptor by inside-out signalling and conformational change of the binding site, or may occur in isolation, as quiescent GPIIb/IIIa maintains a low-affinity for its ligand.
Clustering of integrins is the visible extracellular manifestation of outside-in signalling as, via a variety of ligand- and activation-dependent mechanisms, integrin density increases where soluble ligand is present at the extracellular surface. Clustering activates cytosolic tyrosine kinases (e.g. the Src-family tyrosine kinases via interaction with GPIIIa CT), thus enabling adhesion-dependent protein phosphorylation. 42 The process of clustering is necessary for optimal receptor function in addition to avidity modulation. 43
In addition to this visible process of clustering, arachidonic acid is liberated from the plasma membrane in conjunction with ADP signalling, thus providing substrate for cyclooxygenase (COX) and consequent generation of thromboxane (TXA2). 44
Inhibition of integrin activity
The change in conformation of a GPIIb/IIIa receptor to either the quiescent or active state is not an all-or-nothing phenomenon within an individual platelet, but rather a dynamic equilibrium which is shifted towards one or other state by the interactions of prothrombotic and antithrombotic mediators acting on extracellular ligand-binding sites. Inhibitory signals prevent inappropriate activation and limit thrombus formation to the site of vascular injury.
We have already mentioned that the G-protein-coupled soluble agonists and adhesion receptors act as promoters of the active, prothrombotic state. The molecules primarily responsible for promoting the quiescent state are nitric oxide (NO) and prostacyclin (PGI2), via the generation of cyclic guanosine monophosphate (cGMP) and cyclic adenosine monophosphate (cAMP), respectively. 45
PGI2 is synthesized by inducible COX-2 within the vascular endothelium 46 and is up-regulated in a paracrine fashion by TXA2 released from platelet granules. 47 Elevation of cAMP levels leads to phosphorylation of proteins that inhibit the inside-out activation of GPIIb/IIIa by inhibiting cyto-skeletal re-arrangement. 48
Endothelial NO synthase (eNOS) is integral to platelet function and, with the short biological half-life of NO; its actions are restricted to the immediate vicinity of its synthesis. 49 NO has been found to mediate a number of antiplatelet actions. It inhibits the adhesion of platelets to the endothelium,50,51 suppresses platelet aggregation 52 and stimulates disaggregation. 53
NO acts to inhibit platelet activation via a number of mechanisms which all appear to be cGMP-mediated. 54 In the context of the fibrinogen receptor, cGMP inhibits activation of phosphoinositide 3-kinase (PI3-K), 55 which, via inside-out signalling, normally promotes a conformational change in GPIIb/IIIa that facilitates its binding to fibrinogen. 56 NO also inhibits GPIb-mediated myosin light chain (MLC) phosphorylation and GPIIb/IIIa activation. 57
Dynamic recruitment of fibrinogen receptors
The expression of fibrinogen receptors on the platelet surface is a dynamic not a static process. Alpha granules contain a pool of fibrinogen receptors that are translocated to the plasma membrane on activation.6,58–60 Differential packaging of proteins within alpha granules enables this to be carried out in a selective manner, with translocation of other glycoproteins and secretion of fibrinogen contributing to amplification of signal transduction. 61
The expression of GPIIb/IIIa exhibits a positive relationship with ADP-induced aggregation in both healthy subjects and patients with acute coronary syndromes prior to therapy with oral ADP receptor antagonists. 62 The expression was, however, also found to be related to mean platelet volume, and therefore may not actually represent increased receptor density. A future direction of study should be to investigate whether platelets are able to synthesize GPIIb/IIIa de novo in response to stimuli as is the case with the regulatory protein B-cell lymphoma-3 to control clot retraction. 63
A pathological fibrinogen receptor
The formation of thrombus in response to vascular injury is not due to a pathological fibrinogen receptor per se, as it is an appropriate response to the generation of pro-inflammatory and pro-aggregatory factors released into the intravascular compartment. There are, however, instances when aggregation is enhanced or suppressed due to intrinsic properties of the integrin. We will discuss the common Pro 33 polymorphism, aspirin resistance and GT as examples of these occurrences.
Pro 33 polymorphism in GPIIIa
The platelet fibrinogen receptor is polymorphic with a number of recognized stable allelic variants based on single amino acid substitutions. 64 The GPIIIa Pro 33 polymorphism is one of the more heavily studied variants after it was associated with neonatal alloimmune thrombocytopaenia. A modification of the PlA1 epitope to PlA2 involves an amino acid substitution of proline for leucine at position 33 within the extracellular domain adjacent to the ligand-binding site. Pro 33 has a Caucasian population frequency of 0.15, 65 and in 1996 was noted to cluster with acute coronary thrombosis, especially in patients whose primary event occurred under the age of 60 years. 66
Studies since then have yielded inconsistent results, but overall it has been found that carriers of the PlA2 allele are at increased risk of coronary artery disease (odds ratio [OR], 1.10; 95% confidence interval [CI], 1.03–1.18), with the risk being greatest in those <60 years old (OR, 1.21; 95% CI, 1.05–1.38) and in those with in-stent re-stenosis (OR, 1.31; 95% CI, 1.10–1.56). 67 Morphological studies using magnetic resonance imaging have demonstrated a decrease in the thickness of the fibrous caps covering atherosclerotic plaques 68 in individuals with this polymorphism, and this has previously been associated with increased risk of plaque rupture. 69 Platelet function studies, as with the epidemiological findings, have often yielded contradictory results, but overall the polymorphism appears to associate with increased platelet aggregability 70 and platelet activation. 68
The prothrombotic phenotype seen in Pro 33 individuals is not secondary to an increase in receptor expression but rather appears to be the result of a change in integrin signalling. 71 Data regarding the affinity of fibrinogen binding and alteration of GPIIb/IIIa clustering are inconclusive, but the Pro 33 isoform demonstrates increased serine/ threonine phosphorylation of extracellular signal-regulated kinase and MLC kinase (MLCK). 72
MLCK-mediated phosphorylation of MLCs is necessary for platelet shape change, secretion and the clustering of integrins. 73 Myosin phosphatase (MP) normally dephosphorylates MLCK thus reducing platelet activation. In platelets from individuals with the Pro 33 polymorphism, MP is itself deactivated by phosphorylation resulting in persistent activation of MLCK. 72 The result is ongoing actin re-organization within the platelet via postoccupancy outside-in signalling, and this leads to an increased level of platelet activation and hence increased cardiovascular risk.
Aspirin resistance
Aspirin had been a popular synthetic analgesic agent for almost 90 years before its antiplatelet effects were found to confer a significant survival advantage postmyocardial infarction 74 and in the secondary prevention of cardiovascular disease. 75 A proportion of patients, however, fail to respond appropriately to aspirin in a heterogeneous phenomenon known as aspirin resistance.
The concept of aspirin resistance originated with the clinical observation that a number of patients on aspirin therapy continued to have platelet-mediated thrombotic events. 76 This was perhaps unsurprising considering often suboptimal medication adherence, variable drug pharma-cokinetics and the action of aspirin on only a single pathway of platelet activation. However, it became evident that in some individuals there was a biochemical component of aspirin resistance that could not be overcome with increasing serum levels of the drug. A failure to suppress TXA2 production or to inhibit platelet aggregation in response to the agonist arachidonic acid currently forms the basis of the generally accepted definition for biochemical, or laboratory, aspirin resistance. 77
The reported prevalence of aspirin resistance varies significantly between studies as, despite an accepted biochemical definition,in vitro measurements of platelet function are performed using disparate assays with large inter-assay variability. 78 Two recent systematic reviews have, however, demonstrated some agreement, with the mean prevalence of aspirin resistance being identified as 24% 79 and 28%. 80 In the second study, examination of the relationship of aspirin resistance to clinical events found that a significantly increased proportion of those classified as aspirin-resistant suffered a cardiovascular event (OR, 3.85; 95% CI, 3.08–4.80; P < 0.001). 80 Investigations into the aetiology of this phenomenon have indicated that the fibrinogen receptor, or rather its GPIIIa component, may be involved in many cases.
A number of candidate genes have been investigated as potential causes of aspirin resistance, and the same PlA1/A2 single-nucleotide polymorphism in GPIIIa as described in the previous section has been identified as the strongest candidate in a large systematic review of 50 polymorphisms within 11 genes. 81 The PlA1/A2 polymorphism was found to be significantly associated with aspirin resistance in healthy subjects (OR, 2.36; 95% CI, 1.24–4.49; P =0.009), but not so when combined with patients with cardiovascular disease (OR, 1.14; 95% CI, 0.84–1.54; P = 0.40). The authors of this study identify significant heterogeneity within the studies analysed and suggest caution in the interpretation of their results. However, recent preliminary work from our laboratory analysing the platelet proteome has shown GPIIIa expression to differ between aspirin-sensitive and resistant individuals (unpublished data, Timothy Goodman, 2011).
The data obtained from investigation of the Pro 33 population, many of whom were healthy subjects not taking aspirin, suggest a possible mechanism for aspirin resistance. Oral antiplatelet agents act primarily by dampening the inside-out signalling that leads to GPIIb/IIIa activation. Aspirin achieves this by irreversibly acetylating intracellular COX-1 thereby inhibiting TXA2 production, and the ADP antagonists bind the extracellular ADP receptors thus preventing activation of their G-protein-coupled receptors. The Pro 33 polymorphism appears to result in sustained outside-in signalling yet, as previously stated, aspirin is generally considered to be involved in dampening inside-out signalling. The distinction between signalling direction within the cytoplasm is somewhat of an artificial construct, as many processes are involved in both, and during the process of thrombus formation there is simultaneous outside-in and inside-out signalling. Aspirin is known to acetylate platelet proteins involved in platelet aggregation other than COX-1, and so it is possible that the Pro 33 isoform is more susceptible to acetylation resulting in alteration of function. 82
An alternative hypothesis is that increased intracellular arachidonic acid, the substrate for COX-1, may act to inhibit MP within platelets, as has previously been seen in muscle. 83 This would lead to persistent platelet activation via postoccupancy outside-in signalling as described above.
Studies into aspirin resistance have been handicapped by a combination of small sample size and a lack of standardization in its evaluation and definition. 84 Estimates of prevalence of this phenomenon are still widely debated and, until larger epidemiological studies utilizing a network biology approach to target both putative mechanisms and pharmacogenetics 85 are undertaken, much about this clinically important phenomenon will remain elusive.
Glanzmann's thrombasthaenia
GT was first identified in 1918, and subsequently characterized as a bleeding diathesis involving mucous and cutaneous bleeding in the context of normal platelet count and lifespan, prolonged bleeding time, deficient clot retraction and absent platelet aggregation. 86 The hypothesis that these observations were secondary to deficiencies in the cell membrane was confirmed with the finding of plasma membrane glycoprotein abnormalities.87–90 The phenotype of GT is due to defects/deficiencies in either the alpha or beta subunits of the fibrinogen receptor resulting in either a reduction in receptor expression or, more commonly, expression of a defective receptor. The elucidation of GPIIb/IIIa synthesis, structure and function has occurred in parallel to the investigation of GT and is reviewed in its historical context by Coller and Shattil. 91
Recent platelet aggregation studies have revealed a decreased response to the soluble agonists ADP, arachidonic acid, collagen and epinephrine, but a normal response to ristocetin, 92 in patients with GT. The intracellular pool of fibrinogen within alpha granules is also reduced due to defects in fibrinogen transport and storage.93,94
GT is caused by a diverse set of genetic mutations that are mainly non-sense mutations, out-of-frame and in-frame small deletions and insertions within either the ITGA2B or ITGB3 genes. 95 There is increased prevalence among certain ethnic groups, with a predilection for those populations with a high frequency of con-sanguinity. 96 Over 100 distinct mutations have been identified to date but, due to the low prevalence of specific mutations and significant genetic heterogeneity, it has not yet been possible to assign individual phenotypes. 95 Standard mutagenic techniques are not applicable in nucleated platelets but, by utilizing alternative techniques, knockout models have enabled certain mouse phenotypes to be categorized, many of which are characterized based on detection of the beta-3-mediated defects in the other cell types in which it is expressed. Genetic mutations have been identified throughout the structure of the fibrinogen receptor and result in defects in ligand binding, structural rearrangement, outside-in and inside-out signalling. They are too numerous to elaborate here, but are reviewed elsewhere. 95
Targeting the platelet fibrinogen receptor
In the United Kingdom, almost one-third of deaths are due to cardiovascular disease, with coronary heart disease and stroke accounting for the majority of cases. 97 Coronary thrombosis is the common pathway in transmural myocardial infarction, 98 and platelets are the primary effectors of this. 99
Many different agents to inhibit platelet aggregation have been developed including inhibitors of TXA2 synthesis, ADP receptor antagonists and phosphodiesterase inhibitors. None of these targets however cause complete inhibition of platelet aggregation (IPA),100,101 as individually they each modulate only a single pathway of platelet activation. The potential to inhibit the final common pathway of platelet aggregation may, in contrast, result in more complete and faster IPA. 102
Intravenous GPIIb/IIIa Inhibitors
The intravenous GPIIb/IIIa inhibitors (GPI) were the first rationally designed antiplatelet drugs which act to block the ligand-binding site, thus preventing both outside-in signalling and the aggregatory outcome of inside-out signalling. There are three intravenous GPI that are currently approved for clinical use: Abciximab (a monoclonal antibody fragment), Eptifibatide (a cyclic peptide) and Tirofiban (a peptidomimetic molecule). All three agents act by competing for receptor occupancy with the soluble receptor agonists, and have comparable efficacy in terms of cardiovascular outcomes.103,104
In the context of clinical outcomes, the addition of Abciximab to standard pharmacological therapy during elective coronary stenting, reduced the composite endpoint of death, myocardial infarction and the need for urgent revascularization at both 30 days (hazard ratio [HR], 0.48; 95% CI, 0.33-0.69; P < 0.001) 105 and six months (HR, 0.47; 95% CI, 0.33-0.68; P < 0.001). 106 The mortality benefit is greatest in patients with elevated troponin, 107 and is independent of both coronary lesion morphology 108 and interventional technique. 109 Increased major bleeding is a recognized adverse effect of intravenous GPI, but this can be limited by minimizing the duration of therapy before primary coronary intervention without offsetting its therapeutic effectiveness. 110
Oral GPIIb/IIIa inhibitors
The period soon after myocardial infarction is extremely high risk in the context of platelet-mediated thrombosis. This period of high risk continues for a number of weeks after the event as platelets retain predilection for spontaneous activation.111,112 It would be impractical to maintain patients on intravenous therapy for such a protracted time, and so oral GPI were developed in an attempt to offer prolonged treatment during this period.
Meta-analysis of over 33,000 patients involved in phase III trials of oral GPI demonstrated an increase in mortality in individuals prescribed these agents compared with placebo (OR, 1.37; 95% CI, 1.13–1.66; P = 0.001). 113 This increase in mortality may be due to either toxic effects or a paradoxical increase in platelet activation secondary to partial agonist action due to suboptimal GPIIb/IIIa blockade. 114 The second hypothesis had been previously mooted to explain lack of positive outcomes associated with prolonged Abciximab infusions in the GUSTO IV study, where placebo demonstrated no significant increase in death or myocardial infarction compared with 24 hours (OR, 1.0; 95% CI, 0.83–1.24) or 48 hours (OR, 1.1; 95% CI, 0.94–1.39) Abcixi-mab infusion, at 30 days. 115 Consequentially, the oral GPI (Xemilofiban, Orbofiban and Sibrafiban) were not approved for clinical use.
GPIIb/IIIa inhibitors in clinical practice
The role for GPI in clinical practice is limited, and the current recommendations from the European Society of Cardiology are for intravenous GPI to be used in high-risk primary coronary intervention among patients who are already treated with dual antiplatelet therapy. 116 However, many of the early trials of GPI were without the benefit of either drug-eluting stents or dual antiplatelet therapy, and so the clinical benefits attributed to GPI are likely to be attenuated with the provision of what is now standard therapy. Also, as more potent ADP receptor antagonists and direct thrombin inhibitors are licensed, the role for GPI as an adjuvant antiplatelet agent may diminish.
Despite being the first rationally designed anti-platelet agent, GPI are still a relatively blunt tool as they act by blocking ligand binding rather than modulating signalling. As our understanding of bidirectional signalling evolves, it may prove possible to target specific aspects of outside-in signalling with new agents and this may lead to a possible resurrection of the possibilities for oral therapy.
Therapy for Glanzmann's thombasthenia
In contrast to the prevention of thrombus formation from pathological activation of the fibrinogen receptor, the prevention of bleeding from a pathological lack of appropriate activation is considerably more challenging. Therapy for GT has historically involved multiple platelet transfusions with the associated problems of transfusion antibody formation. The availability of recombinant factor VIIa 117 and bone marrow transplan-tation 118 have been important recent advances in care. Advanced genetic techniques to increase de novo expression of functional fibrinogen receptors are in early phases of development, but offer the potential for cure without the mortality associated with bone marrow transplantation.
Conclusions
The platelet fibrinogen receptor is central to the formation of a platelet-mediated thrombus in response to vascular injury, and is responsible for much pathology via both appropriate and defective responses to stimuli. GPIIb/IIIa synthesis and heterodimer formation are carefully marshalled through organelles designed to transport only functional GPIIb/IIIa to the cell surface. However, the enormous diversity of genetic mutations has resulted in a number of allelic variants becoming established within populations, resulting in the diametrically opposed outcomes of increased cardiovascular risk and bleeding diatheses.
The PlA2 allelic variant and consequent Pro 33 polymorphism in GPIIIa appears to confer increased cardiovascular risk both in the context of increased platelet aggregability and of resistance to aspirin. The increased cardiovascular risk of Pro 33 appears to be less than that for conventional risk factors but, until a large epidemiological study with prolonged follow-up is performed, the true magnitude of the effect will remain unclear. Similarly, regarding the role of Pro 33 in aspirin resistance, larger studies are needed with the added caveat that standardization of the definition of aspirin resistance is required – the subject of aspirin resistance has, since it was first reported, been plagued by under-powered studies using diagnostic tests with high intra- and intersubject variability.
Since the emergence of intravenous GPI and the therapeutic failure of oral GPI, the fibrinogen receptor has been somewhat sidelined as a therapeutic target as the newer, more potent, ADP antagonists and direct thrombin inhibitors have entered clinical practice. As our understanding of the extracellular domain of GPIIb/IIIa and its contribution towards outside-in signalling is further elucidated using modern techniques, the opportunity for more advanced rationally designed GPI may present itself. On the other side, advances in gene therapy and stem cell manipulation may facilitate a cure for GT.
In conclusion, while much has been learnt about the fibrinogen receptor and its contribution to both thrombotic and haemorrhagic pathology, there still remain important questions that will require modern technological methods and large epidemiological studies to answer.