
Abstract
Chronic kidney disease (CKD) is now recognized as a major public health issue. One consequence of this condition is disturbance of mineral and bone homeostasis. Bone disease (renal osteodystrophy) as a consequence of CKD has long been recognized. However, it is now appreciated that the mineral and bone disturbances of CKD (and perhaps treatment of them) lead to vascular calcification, which is a cause of significant morbidity. In recognition of the widespread nature of the condition, the term CKD-mineral bone disorder (CKD-MBD) is now in general use to describe the biochemical, skeletal and vascular changes that occur in CKD. The pathogenesis of CKD-MBD is incompletely understood but has recently been redefined with the emergence of fibroblast growth factor 23 (FGF-23) as a major influence on control of vitamin D and parathyroid hormone. This review describes the classification of CKD and current understanding of the mechanisms underlying CKD-MBD (incorporating FGF-23). It describes and evaluates the means of identifying CKD-MBD in the clinical setting and the interventions available for treatment. It then reviews current clinical guidelines for the use of biochemical markers in clinical decision-making. In acknowledgement of the paucity of evidence upon which these guidelines are based, areas where clinical research might be directed in the future will be identified.
Introduction
Classification of CKD
CKD, chronic kidney disease; GFR, glomerular filtration rate
At least two GFR measurements or estimations must be taken no less than 90 days apart before the patient is classified into one of the stages of CKD 2
The suffix ‘P’ is applied when urine albumin creatinine ratio >30 mg/mmol or protein creatinine ratio >50 mg/mmol
*Other evidence of kidney damage may be one of the following:
Persistent albuminuria Persistent proteinuria Persistent haematuria (after exclusion of urological causes) Structural abnormalities demonstrated by ultrasound, etc
KDIGO definition of CKD-MBD and how it is distinct from renal osteodystrophy (adapted from ref. 4 )
KDIGO, Kidney Disease Improving Global Outcomes; CKD-MBD, chronic kidney disease-mineral bone disorder; PTH, parathyroid hormone
Normal calcium and phosphate homeostasis
In healthy individuals, mineralization of bone and appropriate blood concentrations of calcium and phosphate are maintained by a complex interplay between three feedback loops. The central role of parathyroid hormone (PTH) and vitamin D in this process has long been recognized, but recently, the importance of a third molecule, fibroblast growth factor 23 (FGF-23), has been appreciated. 6 Before we can understand the pathogenesis of CKD-MBD, it is important to review the characteristics of these three molecules and how they interact.
PTH is an 84-amino acid protein synthesized and stored in the parathyroid glands. Once released, it has a half-life of about four minutes. To be cleared from the circulation, it is cleaved into a number of fragments, most (but not all) of which are biologically inactive. These are then metabolized by the liver and kidneys. The extracellular concentration of ionized calcium is the most important determinant of blood PTH. The parathyroid glands express calcium-sensing receptors which stimulate PTH production and secretion in the presence of hypocalcaemia. PTH production is also stimulated by hyperphosphataemia and reduced concentrations of calcitriol. PTH synthesis and secretion are reduced by hypercalcaemia and increased concentrations of calcitriol and FGF-23.
PTH binds to receptors in renal and bone cells and activates a number of secondary messengers. In the proximal tubular cells of the kidney, these deactivate sodium-phosphate co-transporters leading to reduced phosphate absorption. In the bone, PTH has two opposing effects which together increase bone turnover; it stimulates the activity of osteoblasts (increasing bone formation) and increases the number of osteoclasts (increasing bone breakdown). The net effect is the release of calcium and phosphate from bone into the circulation. The phosphaturic effect of PTH on the kidney counteracts the tendency to hyperphosphataemia, so in essence, PTH acts to increase the blood concentration of ionized calcium. This then acts on the calcium receptors in the parathyroid gland to suppress PTH production, thus completing the feedback loop.
Nomenclature of vitamin D2, D3 and their derivatives
The vitamin D metabolites are transported in the blood by a specific binding protein and bind to specific vitamin D receptors expressed at various sites. In the gut and the proximal tubule of the kidney, stimulation of vitamin D receptors increases calcium and phosphate absorption. Calcitriol also directly suppresses PTH secretion (which reduces calcium and phosphate release from bone) and stimulates FGF-23 production (which lowers phosphate absorption).
The effects of vitamin D are controlled by modulation of the 1α-hydroxylation step in the proximal tubular cells of the kidney. This enzyme is stimulated by PTH, calcitonin, low calcium and low phosphate and inhibited by its products (calcitriol and ercalcitriol) and FGF-23, as well as metabolic acidosis. These influences complete another series of feedback loops.
FGF-23 is a circulating 32-kDa peptide secreted by osteocytes, osteoblasts and osteoclasts in bone in response to hyperphosphataemia and increased concentrations of calcitriol. FGF-23 acts via a specific receptor and forms a complex with the transmembrane protein klotho, the latter being a prerequisite for FGF-23 activity. 7 FGF-23 reduces blood phosphate concentration by two mechanisms: it reduces reabsorption of phosphate by the proximal tubule cell in the kidney (antagonizing vitamin D at this site) by acting upon the sodium–phosphate transporter, and it inhibits the enzyme 1α-hydroxylase in the kidney, thus reducing synthesis of calcitriol. 8 The latter has the effect of reducing phosphate absorption in the gut and kidney. The lowered phosphate concentrations then down-regulate FGF-23 production in bone, thus completing the third feedback loop.
In health, the complex interplay between these three feedback loops (Figure 1) maintains blood calcium and phosphate concentrations within tight parameters and ensures appropriate turnover and mineralization of bone. The skeleton can be viewed as a large reservoir of calcium and phosphate which can be called upon to release relatively small amounts of these molecules into the circulation which then act to modulate the hormone-mediated homeostatic responses.
Diagram showing the direct interactions between parathyroid hormone (PTH), calcitriol and FGF-23. These three molecules each have feedback loops which regulate phosphate homeostasis through their respective actions on phosphate absorption from the gut, assimilation into bone and excretion by the kidney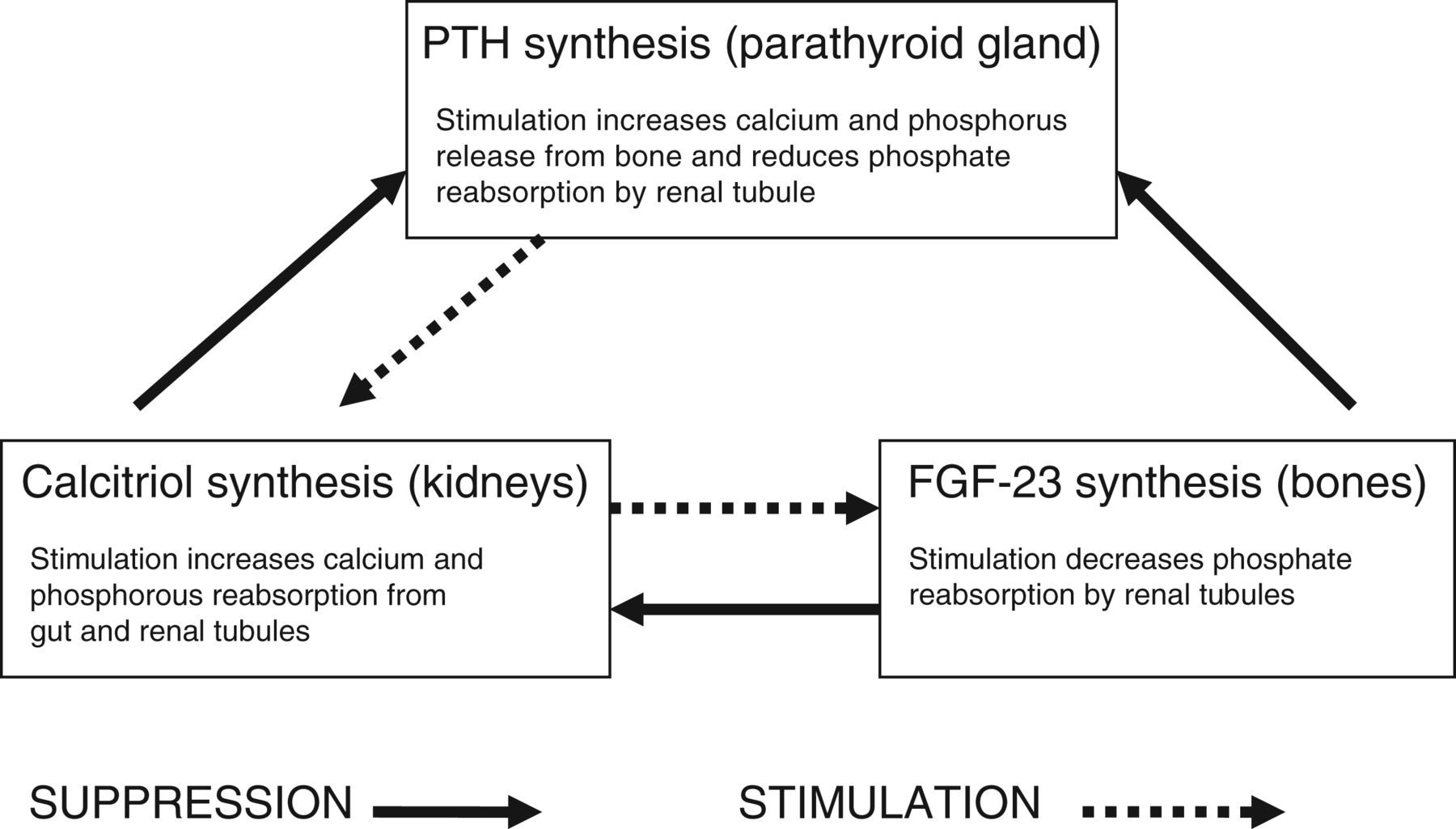
The pathogenesis of CKD-MBD
Although understanding of the mechanism by which CKD disturbs the PTH/vitamin D/FGF-23 axis is incomplete, it is currently accepted that the central defect is the breakdown in the regulatory mechanisms of phosphate homeostasis.
CKD is characterized by glomerular and tubular sclerosis leading to reduced glomerular filtration and reduction in renal parenchymal mass. These changes have two effects relevant to calcium and phosphate homeostasis:
Reduced glomerular filtration of phosphate and impaired tubular responsiveness to hormonal influences leads to phosphate retention; Reduced renal mass leads to reduced activity of 1α-hydroxylase in the renal tubule and thus failure to increase calcitriol production when required.
Measurable hyperphosphataemia as a result of phosphate retention is a relatively late phenomenon in the natural history of CKD, usually becoming clinically evident at stage 4–5 CKD. However, circulating calcitriol concentrations begin to fall at stage 3 CKD either as a direct result of phosphate retention or as a secondary effect via FGF-23 stimulation. Lowered calcitriol leads to reduced calcium absorption from the gut and proximal tubule, thus causing a tendency to hypocalcaemia which is counteracted by increased PTH production and secretion. The net effect is secondary hyperparathyroidism (i.e. abnormally high PTH concentrations as an appropriate response to hypocalcaemia), which further aggravates hyperphosphataemia (positive feedback).
Other relevant effects of progressive CKD include:
Reduced expression and responsiveness of the vitamin D receptor on parathyroid tissue, thus further stimulating PTH production. Circulating calcitriol concentrations can be maintained, but at the expense of increased parathyroid gland activity; Reduced expression of the calcium sensing receptor on the parathyroid gland, which not only affects the normal feedback mechanisms for PTH release, but also stimulates parathyroid hyperplasia; Enhanced parathyroid cell proliferation under direct influence of hyperphosphataemia.
If left uncorrected, the secondary hyperparathyroidism leads to increased mobilization of calcium from bone with bone weakening and a tendency to fracture. Hyperphosphataemia affects osteogenic receptors on smooth muscle cells in the arterial walls which causes them to transform to an osteoblast-like phenotype.
9
This causes calcification (or more appropriately, ossification) of soft tissues, particularly the cardiovascular system. This process is probably compounded by the effect of uraemia on the regulation of recently discovered local inhibitors of calcification which are normally found in vascular smooth muscle.
Under the influences described above, the parathyroid glands gradually undergo hypertrophy and become less responsive to modulatory influences. In late CKD, therefore, PTH production may become autonomous (so-called tertiary hyperparathyroidism) with acceleration of bone destruction and vascular calcification even in conditions which would normally suppress PTH (e.g. hypercalcaemia).
The clinical burden of CKD-MBD
Bone disease
Abnormal quality and quantity of bone can lead to an increased risk of fractures in patients with CKD. Hip fractures are seen approximately twice as often as in patients without CKD. 10 The risk of fracture is increased in patients who have had longer exposure to dialysis. Mortality of patients with CKD-MBD who have a hip fracture is about double that of patients without CKD. 11
The pattern of bone disease in CKD has changed over the last 20 years. In the past, high bone turnover was predominant. Such hyperparathyroid bone disease led to bone deformities in children, short stature and reduced quality of life. Nowadays, the gross skeletal abnormalities (renal rickets) described in the past are exceptionally rare. Instead, there is also a high prevalence (40–70%) of low bone turnover disease, which weakens the bone and increases fracture risk. 12 Such adynamic bone diseases may be the result of an ageing population and the effects of therapy. Effective treatment of CKD-MBD therefore requires attention to both high- and low-turnover bone diseases and aims to steer a course between the two.
Vascular calcification
Extraosseous calcification is very common in patients with advanced CKD. Its prevalence increases with severity of renal dysfunction and the length of time a patient remains on renal replacement therapy. 13 Although calcium may deposit in any soft tissue, it is cardiovascular calcification which has been the focus of most attention.
The arterial calcification associated with CKD-MBD is rather different from that seen in the general population. In the latter, calcification is greatest in the arterial intima where it forms a part of atherosclerotic plaques. These predispose to ischaemic events such as myocardial infarction. In CKD-MBD, there is a greater proportion of calcification in the arterial media which causes vascular stiffness and hypertension. 14
Because available in vivo techniques cannot reliably distinguish between the two patterns of calcification, there is an ongoing debate about the relevance of each to adverse outcomes in CKD patients. Despite this debate, there is little doubt from studies of the outcomes associated with vascular calcification (whatever its distribution) that there is a strong association with cardiovascular events and mortality. 15,16
One other rare form of vascular calcification peculiar to uraemic patients warrants special mention. Calciphylaxis is a condition where small cutaneous blood vessels become calcified, leading to acute, painful necrosis and ulceration of the skin. It is strongly associated with the presence of CKD-MBD, although its exact aetiology remains unclear. It has a high mortality, principally due to overwhelming secondary sepsis.
Diagnosis of CKD-MBD
Although the endpoint of CKD-MBD is bone disease and vascular calcification, clinicians should attempt to predict and pre-empt development of the condition before these features are clinically evident. Biochemical analysis of blood is the mainstay of identifying patients at risk of CKD-MBD and is essential in guiding management.
Biochemistry
The key measurements used in routine screening for CKD-MBD are phosphate, calcium and alkaline phosphatase (ALP). It is recommended in the KDIGO guidelines that these are routinely monitored from stage 3 CKD onwards in adults. Individual measurements should guide management; use of the calcium/phosphate product (which was once thought to be a superior measure of calcium and phosphate homeostasis) is no longer recommended. Monitoring of PTH may also be included at stage 3 CKD, but it should certainly be monitored at stage 4. The frequency of monitoring increases as CKD progresses (see below). It is not routine practice to measure vitamin D concentrations in the UK.
Although the blood level of calcium is tightly regulated in the normal individual, it is less so in patients with CKD who are affected by concomitant therapies (notably phosphate binders) and dialysis. Only the ionized calcium (about 40–50% of total blood calcium) is physiologically active, but measuring this requires total exclusion of air from the sample, making its measurement practically difficult. Instead, the concentration of ionized calcium is estimated from the total calcium by correcting for the serum albumin, which determines the ionized proportion of total calcium. Typically, ‘corrected calcium’ is derived, for example by adding 0.2 mmol/L to the total calcium for every 1 g/L decrease in albumin below 40 g/L. The concentration of calcium in the blood is a poor guide to the presence of underlying CKD-MBD. Although there may be a tendency for hypocalcaemia in CKD, more often homeostatic mechanisms (notably increased PTH secretion) maintain blood calcium concentrations within the normal range.
Phosphate is subject to diurnal and postprandial variation and is affected by dialysis. Unlike calcium, it is largely intracellular, so blood concentrations will be influenced by acidosis and hyperglycaemia. Phosphate concentrations tend to rise as CKD advances and frank hyperphosphataemia is common by stage 4 CKD. Hyperphosphataemia is often the first indicator of underlying CKD-MBD.
ALP is produced predominantly by the liver and bone, but small amounts are also released from the gut, leukocytes and kidney. The source of the ALP cannot be identified during routine measurement, but an immunoassay for bone-specific ALP (b-ALP) is available which, when very high or low, correlates with bone turnover. The additional expense of routinely using this test to monitor response to therapy in CKD-MBD has not been justified. Although total ALP is often raised in states of high bone turnover, there is poor correlation with bone histology and it has not proved useful as a predictor of fracture risk.
The diagnosis of secondary hyperparathyroidism which characterizes CKD-MBD should, by definition, require no more than to identify a sustained elevation of blood PTH. However, studies examining the correlation between PTH concentrations and bone histology, bone turnover, bone volume or fracture rate have given inconsistent results. 17 Very high or low concentrations of PTH do have some predictive value in assessing bone turnover but there is no relationship between PTH concentration and measured bone density.
Measurement of PTH remains a problem. Use of C-terminal and N-terminal assays has been abandoned because reduced clearance of cross-reacting inactive fragments in CKD made the results uninterpretable. An immunoassay for ‘intact’ PTH has been developed and is in widespread use. This too detects C-terminal fragments (thought to be so called 7–84 fragments), especially in patients with CKD. These fragments are antagonistic to intact PTH, so the assay could detect apparently high concentrations of PTH in a situation of hypoparathyroidism at the cellular level. Nonetheless, ‘intact’ PTH measurement is still recommended as the assay of choice. A recently developed ‘whole’ or ‘bioactive’ PTH assay, which avoids the effects of inactive fragments, is available but the clinical utility and predictive value of the test is yet to be established.
Although the routine assays for PTH present problems in interpretation, it is still considered good practice to monitor PTH concentrations in people at risk of, or receiving management for, CKD-MBD. Observation of the trend in PTH concentrations is valuable in interpreting changes in bone turnover and providing a rationale for modification of therapy.
Enzyme-linked immunosorbent assays for measuring FGF-23 are available and are under evaluation. The comparative utility of assays interacting upon the intact molecule or the C-terminal fraction is yet to be established. Although FGF-23 concentrations correlate with those of phosphate, there is a variable relationship with PTH concentrations and no correlation has been established between FGF-23 concentrations and mortality. 18 It is therefore not certain what useful clinical information is imparted when high FGF-23 concentrations are detected. Until the assays have improved and their relationship to the clinical scenario is better defined, measurements of FGF-23 are not considered clinically useful and have no part to play in diagnosis or routine monitoring of CKD-MBD.
Bone biopsy
Bone biopsy can be used to identify a range of bone diseases which are distinguished by rate of turnover, degree of mineralization and bone volume. The prevalence of each pattern of disorder differs in patients with advanced (but undialysed) CKD, peritoneal dialysis and haemodialysis. Adynamic bone disease is particularly prevalent in patients receiving peritoneal dialysis while high turnover states (notably osteitis fibrosa) are more common in patients receiving haemodialysis. Although bone biopsy is the gold standard for diagnosing the pattern of bone disease, it is unpleasant for the patient and its clinical value as a guide to therapy is debatable, particularly in an ageing, increasingly co-morbid population. It therefore does not form part of routine practice in the UK. Nonetheless, bone biopsy is favoured in other countries and KDIGO guidelines state that it is reasonable to undertake this investigation where the nature of the bone lesion is not predictable from analysis of the biochemistry alone.
Radiology
Routine X-ray examination of the skeleton is relatively insensitive for the diagnosis of CKD-MBD. Individuals may have normal X-ray appearances in the presence of advanced histological bone disease and marked disturbance of biochemistry. Accordingly, the presence of obvious X-ray changes should be regarded as evidence of inadequate management. X-rays reveal the extent of advanced CKD-MBD; the hands show resorption of the phalangeal tufts and subperiosteal bone erosion, the long bones may show lucent areas (so-called brown tumours) and the spine may show linear osteosclerosis, giving it a characteristically striped appearance (given the quaint anachronistic term ‘rugger-jersey spine’). X-rays also reveal the extent of vascular calcification. While computed tomography-based techniques are the gold standard for identifying vascular calcification, a lateral X-ray of the abdomen (to identify aortic calcification) is sufficient to identify its presence in most patients with CKD stage 3–5 and is considered the investigation of choice by KDIGO.
Bone density measurements (e.g. DEXA scans) do not reliably distinguish between osteoporosis and CKD-MBD. As CKD is predominantly a disease of the aged, both diseases are likely to be present in the same individual. Furthermore, bone density measurements have little clinical correlation and do not predict fracture risk in CKD patients. They are therefore not recommended for diagnosing CKD-MBD.
Management of CKD-MBD
Summary of therapeutic options for treatment of CKD-MBD
CKD-MBD, chronic kidney disease-mineral bone disorder; PTH, parathyroid hormone
Calcium and phosphate
As phosphate retention is central to the pathogenesis of CKD-MBD, it is logical to take therapeutic steps to limit phosphate intake. Specialist dietetic advice is available in all renal units and a reduced phosphate diet is put into place when the blood phosphate rises above normal. Dairy products are particularly rich in phosphate and most patients with hyperphosphataemia are restricted to only a small amount of milk, cheese and eggs. Other foods which are high in phosphate include beans, nuts, ice-cream and some processed meats. Fizzy drinks such as cola are also high in phosphate. The dieticians usually avoid a blanket ban on phosphate-rich foods, particularly when renal patients may also be restricted in salt, protein and (if diabetic) carbohydrate. Reiterative education, tailored to the individual, regarding which foods to avoid, which to be eaten occasionally and which can be eaten freely is therefore much preferred.
The administration of agents to bind phosphate in food is usually required as CKD progresses. There is much debate as to which agent should be used in this context. Agents containing calcium are inexpensive and well tolerated, but there is a risk that these may contribute to vascular calcification. Non-calcium-containing phosphate binders (lanthanum and sevelamer) have the theoretical advantage of reducing calcium intake and thus slowing vascular calcification. There have been numerous studies examining this putative advantage which have yielded inconsistent results and have not translated into better cardiovascular outcomes. 19 The huge differential in their cost compared with calcium-containing agents has to be borne in mind. Accordingly, in the absence of definitive evidence to the contrary, calcium-containing agents are often used first-line except where vascular calcification is evident. Their use is limited by their tendency to cause hypercalcaemia when used in conjunction with vitamin D analogues.
Aluminium-containing phosphate binders are very effective, but their long-term use has been discouraged in most guidelines. This is because of a link between aluminium toxicity and adynamic bone disease. This relationship came to light at a time when domestic water supplies were clarified using aluminium finings. Dialysis machines use large volumes of domestic water during treatments and this led to many long-term dialysis patients accumulating toxic amounts of aluminium. This caused a very debilitating and deforming form of adynamic bone disease. Although dialysis water now contains only very small concentrations of aluminium, the perception of risk associated with aluminium persists. Whether or not the small amounts of aluminium absorbed from using aluminium-containing phosphate binders poses a clinical risk has not been tested, but in the presence of effective alternatives, their use remains discouraged nonetheless.
KDIGO guidelines recommend that blood phosphate and calcium concentrations should be maintained within the normal range in CKD. Where dietary restriction of phosphate and the use of phosphate binders are insufficient in patients with CKD stage 5, there is some scope to increase the amount of phosphate clearance by intensifying dialysis, although phosphate is not cleared efficiently by these means. The recent introduction of novel home dialysis therapies involving an increase in frequency has been associated with improvement in phosphate control. Manipulation of the calcium content of dialysate can be used to alter calcium balance, but the proportion of body calcium available for exchange by dialysis is tiny, and the effect of this on management of CKD-MBD is likely to be small.
PTH and vitamin D
The optimum PTH concentration in patients with CKD stages 3–5, who are not on dialysis, is not known. As mentioned previously, observation of, and reaction to, trends in PTH concentration is probably more useful than maintaining a demarcated range. Nonetheless, there is observational evidence that extremes of PTH are associated with increased mortality. Accordingly, KDIGO recommends that PTH should be maintained within a range of 2–9 times the upper limit of normal for the assay in use.
An increase in PTH can be expected in the presence of hypocalcaemia or hyperphosphataemia and these should be corrected first. Another stimulus to PTH production is vitamin D deficiency, thus vitamin D analogues are often prescribed early in stage 4 CKD to suppress PTH production. The most commonly used agent in the UK is oral alfacalcidol, the dose of which is titrated to maintain the PTH in the desirable range. Its use is limited by a tendency to cause hypercalcaemia, particularly when used in conjunction with calcium-containing phosphate binders. They can also increase phosphate absorption from the gut, contributing to hyperphosphataemia. Different synthetic analogues may have differing effects on PTH suppression and calcium/phosphate absorption, but studies have not provided irrefutable evidence of this.
Hyperparathyroidism may also be addressed with the use of calcimimetics (cinacalcet is the only licensed example). These drugs augment the signal caused by extracellular calcium on the calcium-sensing receptor in parathyroid tissue, thus directly suppressing PTH release. Studies confirm that cinacalcet reduces PTH concentrations, but evidence of benefit in terms of bone disease, vascular calcification or mortality is currently lacking. 20 Furthermore, treatment with cinacalcet is associated with hypocalcaemia, hyperphosphataemia and an increased requirement for calcium supplements. The long-term consequences of these effects are unknown, thus the use of cinacalcet is recommended only in patients with CKD stage 5. Furthermore, these drugs are expensive and are thus generally restricted to adjunct treatment with vitamin D analogues where the latter have not sufficiently suppressed PTH production.
Correction of acidosis
Since calcium salts in bone might be expected to dissociate in an acid environment, the chronic acidosis of advanced CKD might pose a risk for demineralization of bone or at least predispose to ineffective response to therapy. In fact, the evidence for a benefit in correcting metabolic acidosis is very limited with no randomized control trials (RCTs) in pre-end-stage renal failure patients, none in children and only three small trials in dialysis patients. 21–23 Two of these small trials showed that correction of acidosis with bicarbonate may reduce intact PTH concentrations in dialysis patients, possibly by a direct effect on the parathyroid glands, but the clinical utility of this finding is uncertain. None of the studies were sufficiently powered to show a benefit in clinical outcomes. Further studies to better understand the role of acidosis in CKD-MBD are underway.
Monitoring and titration
Schedule for monitoring biochemical markers of CKD-MBD (KDIGO)
KDIGO, Kidney Disease Improving Global Outcomes; CKD-MBD, chronic kidney disease-mineral bone disorder; PTH, parathyroid hormone
There is continuing debate about the utility of routinely monitoring vitamin D and its metabolites. Measurement of 1,25-dihydroxyvitamin D is not useful because of its fluctuating blood concentrations and short half-life of less than 24 h. The best available assays are those that detect 25-hydroxyvitamin D (calcidol and ercalcidol), which has a half-life of three weeks. Several techniques are in current use and it is important to know which has been used before comparing results. The available techniques are not well standardized and the definition of deficiency is not well validated. This is particularly so because of the effect of latitude on vitamin D concentrations; these are low in individuals from northern Europe, especially in winter, compared with individuals from sunnier areas. It is therefore difficult to use vitamin D monitoring to guide therapeutic decisions. Accordingly, monitoring of vitamin D concentrations is not routine practice in the UK outside clinical trials or research projects.
Although the effects of CKD-MBD are detectable in skeletal X-rays, bone density measurements and bone histology, the clinical utility of these investigations as a method of guiding therapeutic decision-making is questionable. These are not generally used for routine monitoring of patients with CKD-MBD.
In the clinical setting, maintenance of biochemical equilibrium in CKD-MBD is difficult. Diet, dialysis regimen and the administration of phosphate binders, vitamin D analogues and calcimimetics each have confounding effects; for instance, an increase in the dose of alfacalcidol in order to suppress PTH may augment intestinal phosphate absorption, necessitating an increase in phosphate binders which (if these are calcium-containing) may lead to hypercalcaemia requiring a change of binders; and if the alfacalcidol is ineffective, cinacalcet may be added which may then cause hypocalcaemia, necessitating further revision of alphacalcidol and calcium supplement doses. Management of the biochemical manifestations of CKD-MBD can best be described as juggling and tweaking; only with regular measurement of the important biochemical markers (phosphate, calcium, ALP and PTH) can rational therapeutic interventions be made.
Outcomes from treatment
Although the management schedule described in this review is predicated on our current understanding of the pathophysiology of CKD-MBD, evidence of its beneficial impact on important outcomes is circumstantial if it is there at all. For instance, observational data have shown that hyperphosphataemia is associated with increased mortality and there is evidence that intervention can lower blood phosphate concentrations, but there are no trials showing that reduction of phosphate reduces mortality. Similarly, cinacalcet reduces PTH, but its therapeutic impact on vascular calcification, fracture rate and mortality is currently unknown.
A review of studies on the outcome of treatment for CKD-MBD undertaken during preparation of the KDIGO guidelines showed that of 15 studies of patient-centred outcomes (mortality, cardiovascular disease, hospitalization and progression of CKD, quality of life and fracture rate), none of them was graded as high quality; three were moderate quality and 12 were low quality. Most studies hitherto undertaken have used biochemical surrogates and have inferred a clinical correlate upon which to base treatment recommendations.
The absence of robust evidence of benefit from intervention was acknowledged by KDIGO, who used a ranking system in their guidelines to distinguish recommendations based on good-quality data from those based on little more than expert opinion or supposition. The striking observation from reading these guidelines is that most of them are based on evidence graded as moderate or poor – and many are based on no trial evidence at all. Clearly, there is a dire need for good-quality research. This is particularly so since some of the treatments on offer (notably cinacalcet and non-metallic phosphate binders) are very expensive; it is important to know if funding these agents translates into improved welfare for patients.
Key areas of future research
The fundamental gap in our current knowledge is whether or not the biochemical abnormalities associated with CKD-MBD can be used to predict the risk of harm (i.e. bone fractures due to bone weakening, cardiovascular events due to vascular calcification, hospitalization and reduced quality of life or death from any cause) and if correction of these biochemical abnormalities reduces this risk.
The first required step is to define normal and pathological values for important variables in the specific context of CKD at its various stages. This might be achieved with a prospective study to determine if measurable biomarkers can be used to predict risk. The relationship between cardiovascular calcification and mortality needs to be better understood and the role of screening for calcification in identifying patients at risk needs to be evaluated. There is a need for a placebo-controlled prospective comparison of different phosphate binders to assess their differential effects on calcification and cardiovascular mortality. The effects of treatment with calcitriol and cinacalcet on these outcomes also need to be determined. The RCTs need to be of sufficient length of follow-up to determine hard outcomes rather than surrogates such as biochemical, histological or radiological effects.
Conclusion
Although the relationship between CKD and bone disease has been established for many years, the exact pathogenesis of CKD-MBD remains poorly defined. The diagnosis of the condition relies heavily on serial analysis of blood biochemistry and management depends on therapeutic intervention when adverse trends are observed. CKD-MBD is associated with adverse outcomes in patients and the burden of morbidity justifies attempts to treat it. Even though the various interventions seem rational in the context of our current understanding of the pathophysiology of CKD-MBD, robust trial evidence of benefits is sparse. There is a great need for research in this area not only to optimize intervention, but also to justify the resources used for current treatment practices.
DECLARATIONS