
Abstract
Background
Accurate measurement of haemoglobin A1c (HbA1c) is useful for long-term glycaemic control in patients with diabetes. Many Hb variants can interfere with HbA1c measurement and cause inaccurate results.
Methods
The subject was a 31-year-old Thai man who was discovered because of an unexpected HbA1c result; other diabetic parameters were within the normal range. Abnormal Hb was investigated using automated high-pressure liquid chromatography (HPLC) and a capillary electrophoresis system. Mutation analysis was done by cDNA sequencing, polymerase chain reaction–restriction fragment length polymorphism (PCR–RFLP) and multiplex allele-specific PCR assays.
Results
Evaluation of HbA1c by cation-exchange HPLC showed a value of 34.9% (reference interval, 4.0–6.0%), but a value of only 4.0% (reference value, 4.8–5.9%) was found with a turbidimetric immunoassay. Haematological analysis revealed a mild anaemia but other parameters were within the normal range. Hb-HPLC analysis demonstrated an unknown Hb variant (47.0%) separating from HbA (46.7%), but capillary electrophoresis identified no abnormal peaks. Mutation analysis identified the Hb Raleigh (β1[NA1]Val → Ala [G
Conclusions
Hb Raleigh can cause falsely increased HbA1c values on cation-exchange HPLC. Definitive diagnosis of this variant using combined Hb and DNA analyses is therefore essential.
Introduction
Haemoglobin A1c (HbA1c) or glycated Hb is produced by a non-enzymatic addition of a glucose molecule to the N-terminal valine residue on the β-globin chain of HbA. Its use historically has been primarily for monitoring long-term glycaemic status. It reflects the average blood glucose concentration during the preceding two to three months and increased HbA1c concentration is a valuable indicator for long-term diabetic control. 1–4 HbA1c assays can be divided into methods based on molecular charge (e.g. high-pressure liquid chromatography [HPLC] and electrophoresis) and methods that use molecular structure (e.g. immunoassays, affinity chromatography and mass spectrometry). Structural Hb variants and the chemically modified Hb derivatives may interfere with HbA1c measurements by these methods. 5,6 Differential diagnosis of Hb variants during HbA1c evaluation is needed for an accurate HbA1c result. We report an interaction between a β-globin chain variant, namely Hb Raleigh (β1[NA1]Val → Ala) 7 and α +-thalassaemia, found in an adult Thai individual who had an unusually high concentration of HbA1c on cation-exchange HPLC but a normal concentration with an immunoassay. The molecular and haematological characteristics associated with this hitherto undescribed combination, as well as differential diagnosis using simple DNA analysis, are described.
Materials and methods
Subject, haematological and biochemical analyses
This study was approved by the Institutional Review Board of Khon Kaen University (HE510728) and informed consent was obtained. The subject, a 31-year-old Thai man, and his pregnant wife were first investigated at the Regional Health Promotion Center 5, Nakhon Ratchasima, Thailand for thalassaemia and haemoglobinopathies as they both had positive screening results for these conditions using a combined mean corpuscular volume (MCV) and dichlorophenolindophenol test.
8
Haematological parameters were recorded using a standard blood cell counter. Ethylenediaminetetracetic acid blood from the subject was transferred to Khon Kaen University for further analysis of a suspected unknown Hb variant. Hb analysis was performed using an automated HPLC analyser (VARIANT™; Bio-Rad Laboratories, Hercules, CA, USA) and capillary zone electrophoresis (Capillarys 2; Sebia, Lisses, France)
9
as shown in Figure 1. Determination of HbA1c level was performed using cation-exchange HPLC (D-10™; Bio-Rad Laboratories,) and a turbidimetric inhibition immunoassay on the Cobas C 501 analyser (Tina-quant™ Haemoglobin A1c Gen.2; Roche Diagnostics, Indianapolis, IN, USA).
Haemoglobin (Hb) analysis of the patient using automated high-pressure liquid chromatography (a) and capillary electrophoresis system (b). Hb Raleigh, HbA and HbA2 are indicated. Note the co-separation of Hb Raleigh and HbA on the capillary electrophoresis system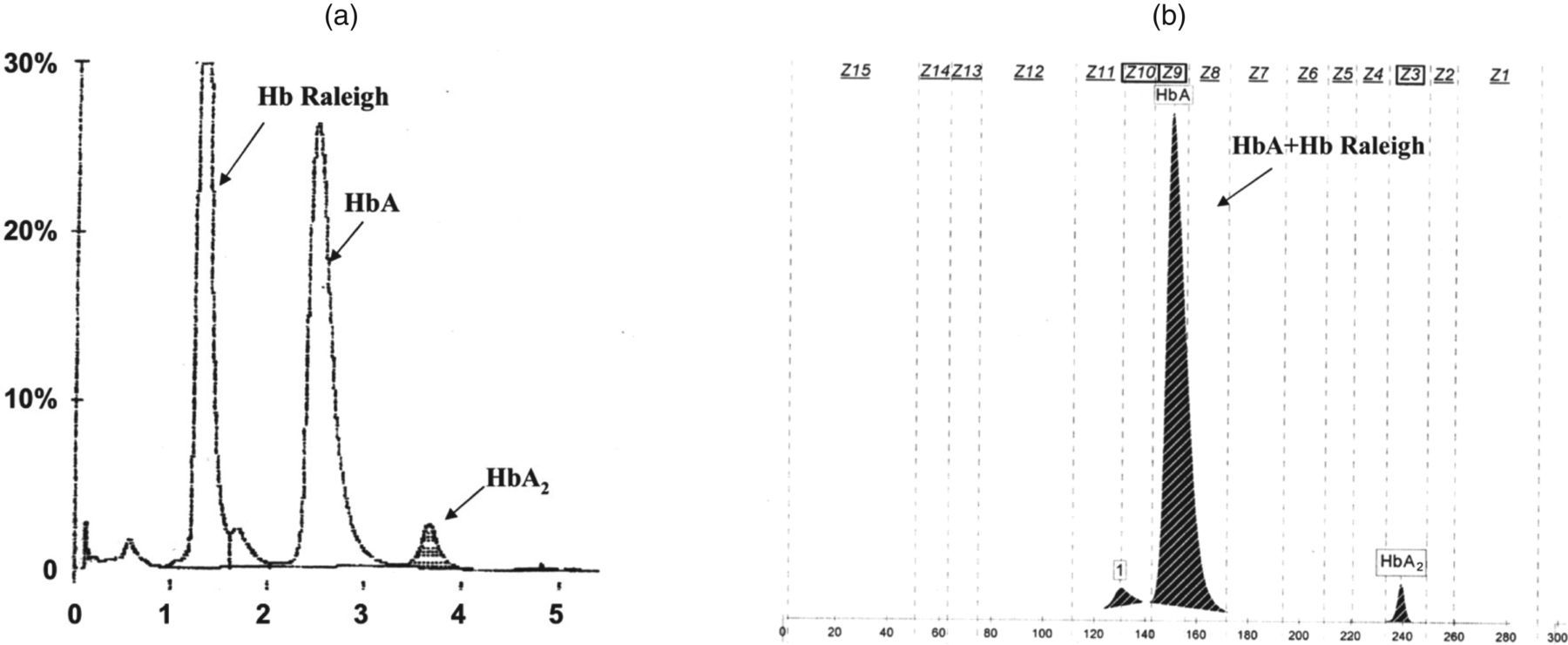
Globin cDNA analysis
Total RNA was isolated from the erythrocytes of the patient using a total RNA blood kit (PureLink™; Invitrogen Corporation, San Diego, CA, USA). Reverse transcriptase polymerase chain reaction (RT-PCR) for each globin gene was performed on a one step RT-PCR kit (MyTaq™; Bioline Ltd, London, UK) using specific globin gene-specific primers.
10
Sequencing of the amplified cDNA was performed with an ABI PRISM™ 3130 XL analyser (Applied Biosystems, Foster City, CA, USA) (Figure 2a).
The anti-sense strand β-globin cDNA sequencing profile of the patient demonstrating a G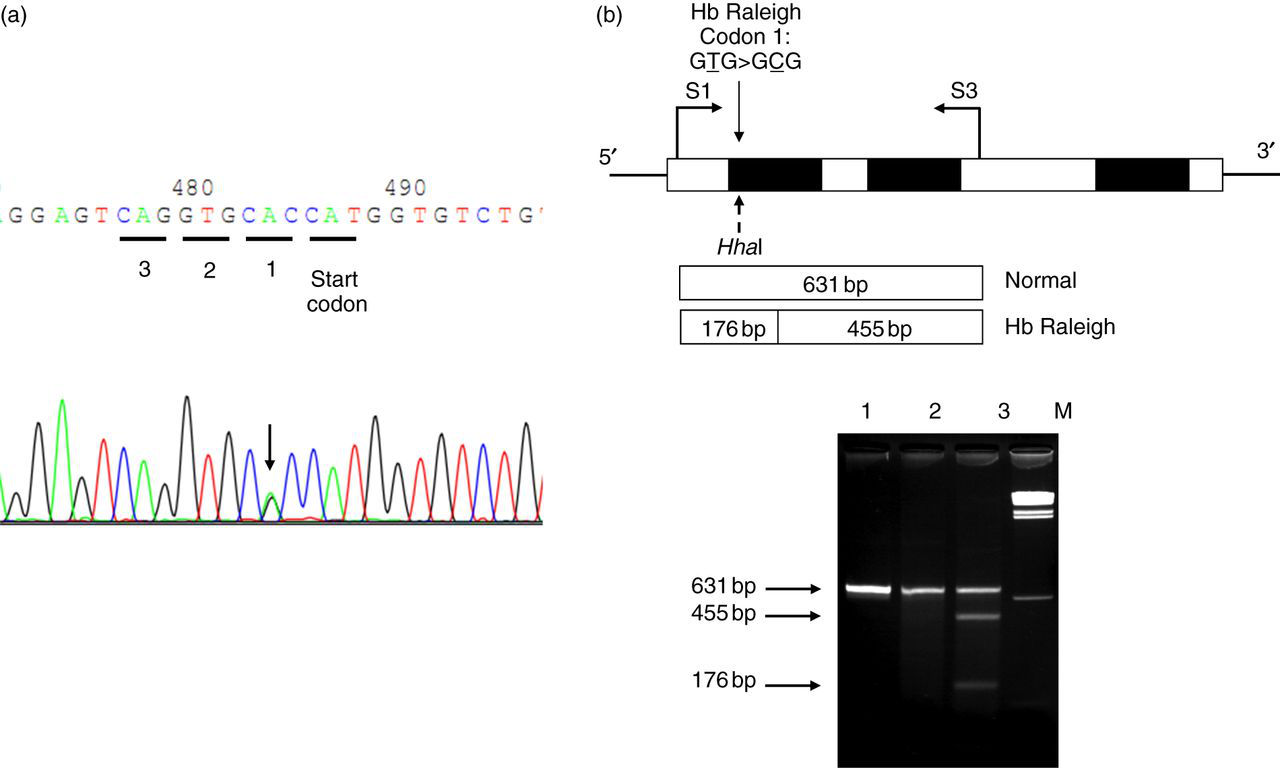
DNA analysis and differential diagnosis of Hb Raleigh and other β-chain variants
Genomic DNA was extracted from the peripheral blood leukocytes. Screening for common thalassaemia genes in Thailand, including αo-thalassaemia (SEA and THAI deletions), α+-thalassaemia (3.7 and 4.2 kb deletions), Hb Constant Spring and Hb Paksé, were routinely performed in our laboratory using PCR-based methods as described elsewhere. 11,12 Identification of similar Hb variants found in Thailand, including Hb Hope (β136[H14]Gly → Asp), Hb Phimai (β72[E16]Ser → Thr), Hb Hekinan (α27[B8]Glu → Asp) and Hb Nakhon Ratchasima (α63[E12]Ala → Val), were carried out using allele-specific PCR methods as described. 13–16 The PCR-restriction fragment length polymorphism (PCR-RFLP) assay for detection of Hb Raleigh mutation on the β-globin gene was developed as shown in Figure 2b. Amplification was done using primers S1 (5′-TGTCATCACTTAGACCTCAC-3′) and S3 (5′-TCCCATAGACTCACCCTGAA-3′) and the amplified product was digested to completion with HhaI restriction enzyme (5′-GCG▾C-3′) (New England Biolabs, Inc., Beverly, MA, USA). The 631 bp Hb Raleigh-derived fragment was digested into two fragments of 176 and 455 bp in length.
To provide a rapid method for differentiation of Hb Hope, Hb Phimai and Hb Raleigh, a multiplex allele-specific PCR was developed as shown in Figure 3. With this system, the primer S3 was used to produce specific fragments with two forward primers including G64 (5′-AAGTGCTCGGTGCCTTTAC-3′) for Hb Phimai and G75 (5′-CCTCAAACAGACACCATGGC-3′) for Hb Raleigh. Primers G41 (5′-TATCAGAAAGTGGTGGCTGA-3′) and H5 (5′-GCAGCCTCACCTTCTTTCATGG-3′) were used to identify the Hb Hope mutation. In the same reaction, two additional primers, γ4 (5′-GGCCTAAAACCACAGAGA-3′) and γ5 (5′-CCAGAAGCGAGTGTGTGGAA-3′), were added for amplification of the G
γ-globin gene promoter, an internal control of the system. The expected fragments of internal control, Hb Raleigh, Hb Hope and Hb Phimai in this multiplex allele-specific PCR were 578, 473, 333 and 131 bps, respectively. A multiplex PCR reaction mixture (50 μL) contained 50 ng genomic DNA, 30 pmol of primers γ4, γ5, H5, S3, G41, G64 and G75, 200 μmmol/L deoxynucleotide triphosphates and one unit Taq DNA polymerase (New England Biolab, Inc.) in 10 mmol/L Tris-HCl (pH 8.3), 50 mmol/L KCl, 0.01% gelatin and 3 mmol/L MgCl2. The amplification reaction was carried out on a T-Personal thermocycler (Biometra GmbH, Gottingen, Germany). A total of 30 cycles after initial heating at 94°C for three minutes was performed under the following PCR conditions: 94°C for 45 s, 65°C for one minute and 72°C for 45 s. The amplified product was analysed on 1.5% agarose gel electrophoresis and visualized under ultraviolet light after ethidium bromide staining.
A multiplex allele-specific polymerase chain reaction for rapid differentiation of haemoglobin (Hb) Hope, Hb Phimai and Hb Raleigh mutations. The locations and orientations of primers (G41 and H5), (G64 and S3) and (G75 and S3) that produce fragments of 333, 131 and 473 bp specific for Hb Hope, Hb Phimai and Hb Raleigh mutations are indicated. The 578 bp band is an internal control fragment of the G
γ-globin gene promoter. Lane 1, normal control; lane 2, Hb Phimai carrier; lane 3, Hb Hope carrier; lane 4, the patient with Hb Raleigh/α
+-thalassaemia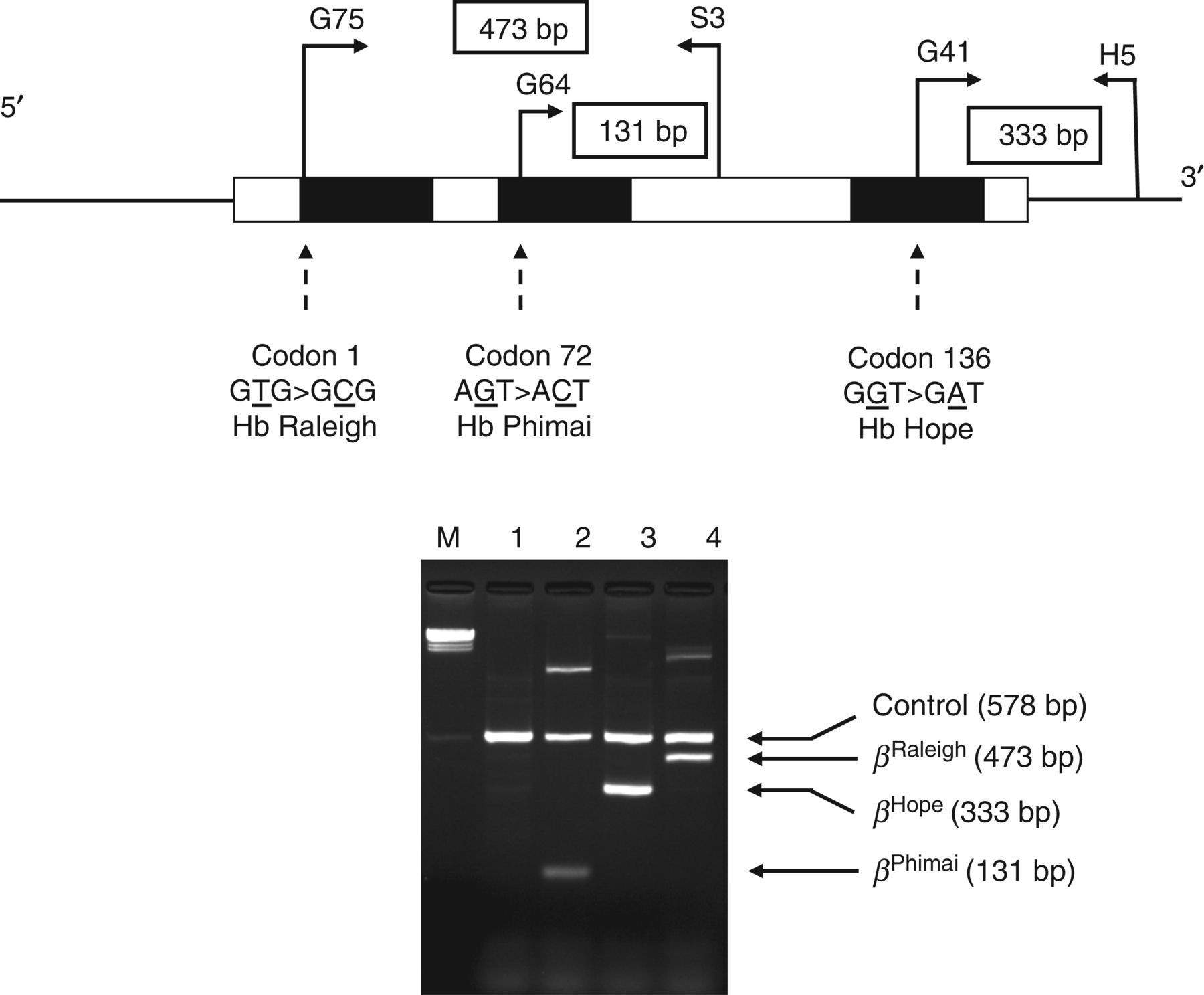
Results
The subject was discovered by our thalassaemia screening programme. Complete blood count revealed mild anaemia with red blood cells 4.0 × 1012/L, Hb 11.8 g/dL, haematocrit 38.0%, MCV 94.8 fl, mean corpuscular haemoglobin 29.4 pg and mean corpuscular haemoglobin concentration 31.1 g/dL. At routine check-up, an initial HbA1c determination by cation-exchange HPLC (Bio-Rad D-10™; Bio-Rad Laboratories) showed a concentration of 34.9% (reference interval, 4.0–6.0%). He had no history of diabetes mellitus and other blood chemistry parameters including blood glucose were within normal range. Re-examination of glycated Hb using a turbidimetric inhibition immunoassay on the Cobas C 501 analyser (Roche Diagnostics) was performed that revealed a concentration of 4.0% (reference interval, 4.8–5.9%). These unusual HbA1c results prompted us to investigate if this was due to potential haemoglobinopathy. As shown in Figure 1a, Hb analysis using HPLC showed 46.7% HbA, 3.0% HbA2 and a peak of an unknown variant at P2-window (47.0%). However, analysis using the capillary electrophoresis system demonstrated a normal Hb pattern with 94.6% HbA and 2.8% HbA2 (Figure 1b), the data indicating the possibility of mis-diagnosis of this Hb variant with the latter. The presence of this Hb variant suggested that the abnormal HbA1c result obtained with the cation-exchange HPLC could be due to the elution of this Hb variant, which has a retention time similar to that of HbA1c.
This Hb analysis result indicated that the patient was heterozygous for an unknown β-globin chain variant with a similar HPLC profile to those of Hb Hope (β136[H14]Gly → Asp), Hb Phimai (β72[E16]Ser → Thr), Hb Hekinan (α27[B8]Glu → Asp) and Hb Nakhon Ratchasima (α63[E12]Ala → Val) found in our series.
13–16
However, allele-specific PCR assays later ruled out all these Hb variants (data not shown). Further β-globin cDNA analysis by DNA sequencing identified the G
Discussion
HbA1c is formed by the non-enzymatic linkage of a glucose molecule to the N-terminal valine residue of the β-globin chain of HbA. The rate of formation of HbA1c is directly proportional to the glucose concentration in the blood. Because of this, accurate HbA1c concentration has been recommended as an indicator of long-term glycaemic control in patients with diabetes. 4 Methods that have been employed to determine HbA1c use separation based on charge differences (chromatography, HPLC or electrophoresis) or structural differences (affinity chromatography or immunoassay).
We have demonstrated in this study that the commonly used cation-exchange HPLC HbA1c assay (D-10™; Bio-Rad Laboratories) can report a falsely high value of HbA1c in the presence of haemoglobinopathy. Others have also noted the same findings. 18–20 The patient was a double heterozygote for Hb Raleigh (β1[NA1] Val → Ala) and an α +-thalassaemia (3.7 kb deletion). He was generally healthy but had a mild anaemia due to the α +-thalassaemia. An amino acid valine at codon one of a normal β-globin chain provides the most common site of glycation within the Hb tetramer. Replacement of valine at this position with alanine in Hb Raleigh can lead to a post-translational modification by acetylation, preventing glycation of the Hb Raleigh molecule. 18 The finding of a relatively low proportion of HbA1c (4.0%) by immunoassay in the patient indirectly supports this. However, this amino acid substitution results in a charge difference similar to glycation, making the behaviour of Hb Raleigh identical to HbA1c on the cation-exchange HPLC method, resulting in a falsely high HbA1c value as measured using cation-exchange HPLC. This and other observations confirm that the Hb Raleigh, either in heterozygote form or in combination with other haemoglobinopathies, could cause a spuriously high HbA1c result on cation-exchange HPLC assay. 18–20 Although the level of HbA1c is unaffected on the immunoassay, laboratory staff should suspect haemoglobinopathy when a spurious HbA1c result is obtained, especially when it is discordant between assays, or with the clinical history of the patient. In this case, selection of an appropriate HbA1c method is essential to provide an accurate HbA1c result and further identification of the suspected Hb variant by both Hb and DNA analyses is preferable.
Hb Raleigh is relatively rare, found mainly among Europeans and could be considered as a clinically silent variant without pathological manifestations. Nevertheless, differentiation from other clinically overt haemoglobinopathies is still essential. Identification of Hb Raleigh in our Thai subject indicates that this Hb variant may not be uncommon as has been thought previously. 21 As shown in Figure 1, laboratory diagnosis of Hb Raleigh may be problematic in a routine setting using a capillary electrophoresis system. In contrast, although HPLC can facilitate its recognition, diagnosis is possible only by means of DNA analysis. Hb analysis using combined assays may be a practical alternative for making presumptive diagnosis, 9,22 with DNA analysis providing definitive diagnosis. The approach we have described here (PCR-RFLP assay using HhaI restriction digestion and a multiplex allele-specific PCR assay demonstrated in Figures 2 and 3 for the detection and differentiation of Hb Raleigh) should prove useful in complementing routine Hb analysis for a definitive diagnosis.
DECLARATIONS