
Abstract
The term amyloid describes the deposition in the extracellular space of certain proteins in a highly characteristic, insoluble fibrillar form. Amyloidosis describes the various clinical syndromes that occur as a result of damage by amyloid deposits in tissues and organs throughout the body. The clinical significance of amyloid varies enormously, ranging from incidental asymptomatic deposits to localized disease through to rapidly fatal systemic forms that can affect multiple vital organs. Currently available therapy is focused on reducing the supply of the respective amyloid fibril precursor protein and supportive medical care, which together have greatly improved survival. Chemotherapy and anti-inflammatory treatment for the disorders that underlie AL and AA amyloidosis are guided by serial measurements of the respective circulating amyloid precursor proteins, i.e. serial serum free light chains in AL and serum amyloid A protein in AA type. Quality of life and prognosis of some forms of hereditary systemic amyloidosis can be improved by liver and other organ transplants. Various new therapies, ranging from silencing RNA, protein stabilizers to monoclonal antibodies, aimed at inhibiting fibril precursor supply, fibril formation or the persistence of amyloid deposits, are in development; some are already in clinical phase.
Introduction
Classification of systemic amyloidosis by precursor protein
DRA, dialysis-related amyloidosis
Fibril formation and amyloid proteins
Amyloid fibrillogenesis remains poorly understood. Experiments have shown in vitro that nearly any polypeptide chain can be driven towards misfolding and aggregation given specific conditions, 4 but relatively few proteins are amyloidogenic in vivo. The polypeptides involved in amyloidosis are structurally diverse in their normal conformation and may be notably rich in β-sheet, α-helix or β-helix. 5 During amyloidogenesis, multimeric proteins dissociate to their monomeric components, and may further be enzymatically cleaved before or during their conversion into amyloid fibrils. 6,7 There are essentially three circumstances in which amyloid deposition occurs. The first is when there is sustained abnormally high abundance of certain proteins that are normally present at low levels, such as serum amyloid A protein (SAA) in chronic inflammation, underlying susceptibility to AA amyloidosis. The second is when there is normal abundance of a normal, but to some extent inherently amyloidogenic protein over a very prolonged period, such as transthyretin in senile amyloidosis (ATTR). The third situation is the presence of an abnormal protein with markedly amyloidogenic structure, such as certain monoclonal immunoglobulin light chains in AL amyloidosis and genetic variants of transthyretin, apolipoprotein AI and fibrinogen Aα chain, etc. in hereditary amyloidosis. Despite the heterogeneity of the various precursor proteins, the morphological structure and histochemical properties of all amyloid fibrils are remarkably similar. The core structure comprises anti-parallel β-strands of polypeptide chains lying perpendicular to the long axis of the fibril. 8 On electron microscopy, amyloid fibrils are characteristically straight, non-branching and 7–10 nm in diameter. 9 Amyloid deposits also contain the non-fibrillar normal plasma protein, serum amyloid P component (SAP), 10 which is bound in a reversible calcium-dependent manner to a ligand present on all amyloid fibrils. Binding of SAP stabilizes the amyloid fibril and protects it from degradation by proteases and phagocytic cells in vitro. 11 Amyloid deposits also contain several other common constituents such as heparan sulphate and dermatan sulphate proteoglycans and glycosaminoglycans, apolipoprotein E, type IV collagen and laminin. Glycan molecules may also contribute to the stabilization of the fibrillar conformation, and may also promote fibrillogenesis. 12
Although there is no doubt that substantial amyloid deposits disrupt organ function through their physical presence, it remains possible that pre-fibrillar amyloid aggregates may also have toxic effects, a hypothesis that has so far mostly been explored with respect to the heart. 13
Diagnosis of amyloidosis and imaging
Amyloidosis is extremely heterogeneous and clinical presentation varies widely depending on which organs are involved. Diagnosis is often made late in the course of the disease, and frequently as an unexpected histological finding when a failing organ is biopsied. Congo red staining of tissue yielding the characteristic apple green birefringence under crossed polarized light remains the gold standard for confirming the presence of amyloid, this pathognomonic optical effect being produced by alignment of the dye molecules along the fibrils (Figure 1). The protein composition of the amyloid fibril, i.e. the type of amyloidosis, must then be ascertained, and this is most accessibly achieved by immunohistochemistry. However, immunohistochemical staining of amyloid deposits can be confounded by many factors including background staining and loss of antigenic determinants in the fibrillar conformation, especially in the common AL type. Proteomic analyses comprising mass spectrometry on amyloid material cut out from tissue sections by laser capture microscopy has lately proved to be effective in a large proportion of cases.
14–16
Whereas target organ biopsies are usually diagnostic, amyloid deposition can be patchy and random ‘screening’ biopsies, e.g. of the gut, are negative in a considerable proportion of cases.
(a) Renal biopsy showing characteristic histological appearance of amorphous amyloid deposits stained with Congo red. This specimen shows predominant deposition within the glomerulus in a patient with hereditary fibrinogen amyloidosis. (b) Same section viewed under crossed polarized light demonstrating apple green birefringence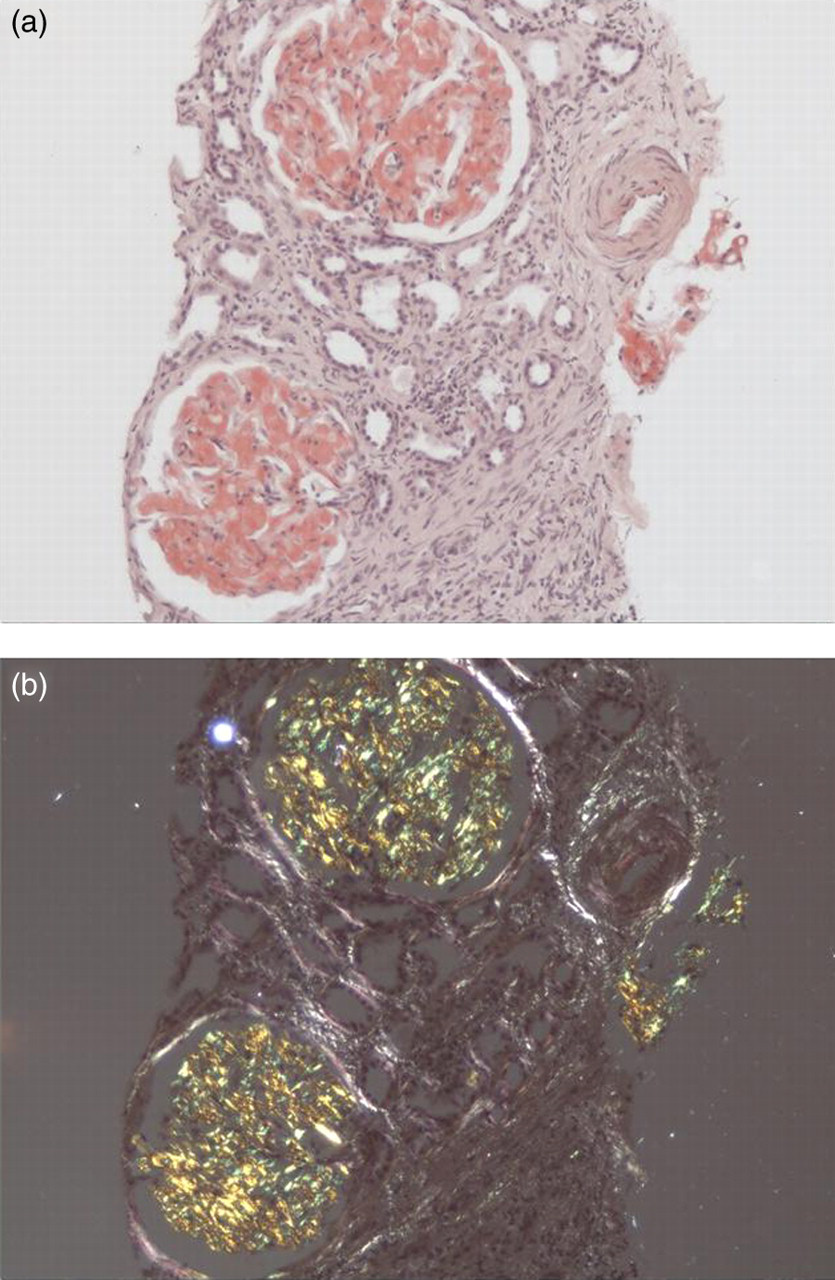
Various imaging techniques can make an important contribution to the diagnosis and evaluation of organ involvement in amyloidosis. Of these, only radiolabelled SAP scintigraphy is specific.
17
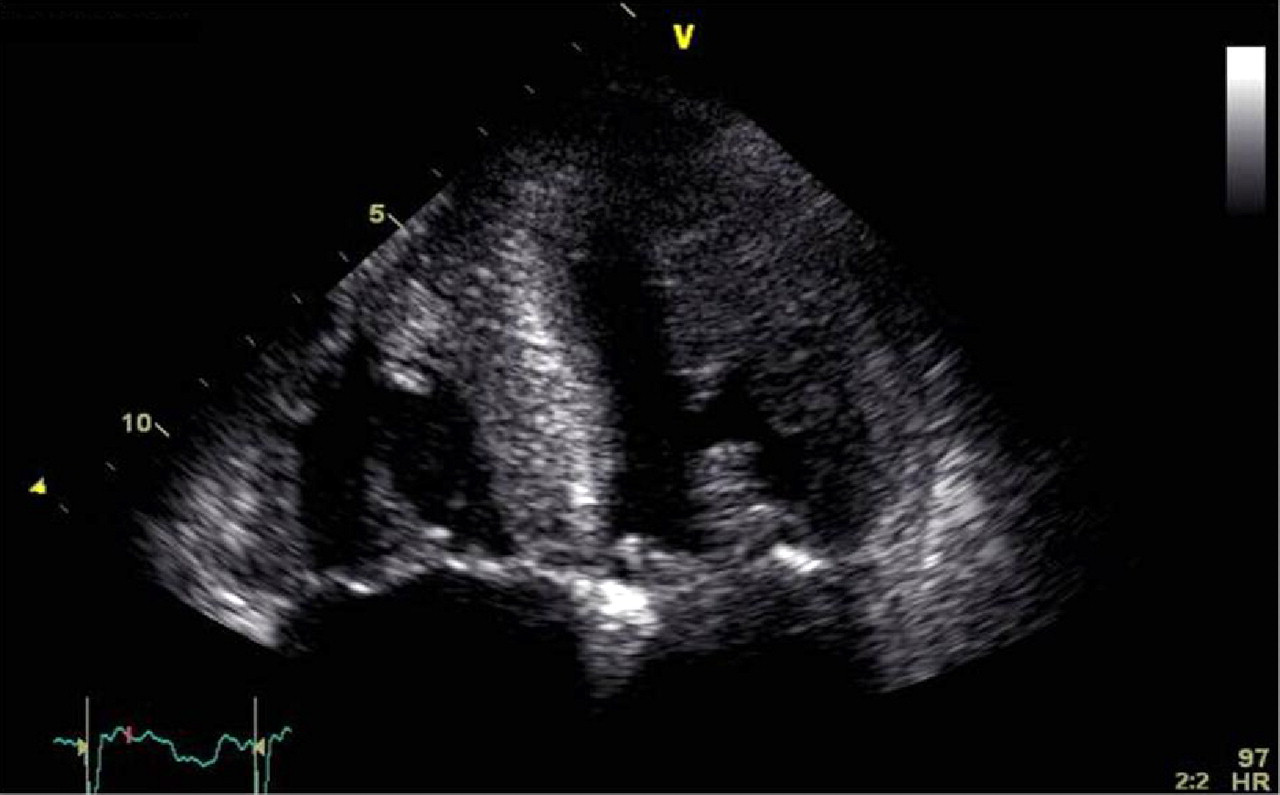
The normal plasma protein SAP binds reversibly to all types of amyloid fibril, and is present in all amyloid deposits. Intravenous injection of 123I-SAP rapidly equilibrates between the relatively small quantity of endogenous SAP within the circulation and SAP present within the extravascular amyloid deposits. SAP scintigraphy thus enables visceral amyloid to be imaged for diagnostic purposes, and since the method is quantitative, it enables the deposits to be monitored serially. SAP scintigraphy has characterized the dynamic nature of amyloid, and has shown that frequently the deposits gradually regress when the supply of the respective precursor protein is reduced (Figure 2). Limitations include restricted availability, and the inability to image amyloid deposits in small or moving structures such as nerves and the heart. Fortunately, several cardiac imaging modalities yield characteristic and clinically important information regarding myocardial involvement. Echocardiography has long been used to demonstrate thickening of the ventricular walls and valves, and to evaluate the predominant diastolic restrictive abnormality that occurs in cardiac amyloidosis (Figure 3). Cardiac involvement has been defined as a mean left ventricular wall thickness of >12 mm in the absence of hypertension or other causes of left ventricular hypertrophy,
18
although poor echocardiographic windows and interoperator variability are significant limitations. Cardiac magnetic resonance (CMR) imaging has lately fallen into widespread clinical practice, demonstrating very characteristic late gadolinium enhancement in subendocardium or more diffusely.
19,20
Although the role of CMR for monitoring progression or regression of amyloid has yet to be defined, the use of equilibrium CMR may prove to be a useful tool in quantification of amyloid, a technique which has been validated in fibrosis.
21
The biomarkers N-terminal pro brain natriuretic peptide (NT-proBNP) and cardiac troponins are also now widely used to provide information on cardiac involvement, prognosis and response to chemotherapy in AL amyloidosis.
22
(a) Anterior whole-body scintigraphic image following intravenous injection of 123I-human serum amyloid P component in a patient with AL amyloidosis at diagnosis. Uptake is seen in the liver, spleen and bones. (b) Same patient as figure (a), following treatment five years later. Marked regression of amyloid deposits can be seen with a small load now seen in the liver and equivocal uptake in the spleen. (c) Posterior scintigraphic image of a patient with AA amyloidosis. Uptake is seen in the spleen. (d) Same patient as figure (c), 10 years later showing progression of amyloid deposits in the spleen Echocardiographic image from a patient with severe cardiac AL amyloidosis. Apical four-chamber view showing massive thickening of the interventricular septum and increased echogenicity of the myocardium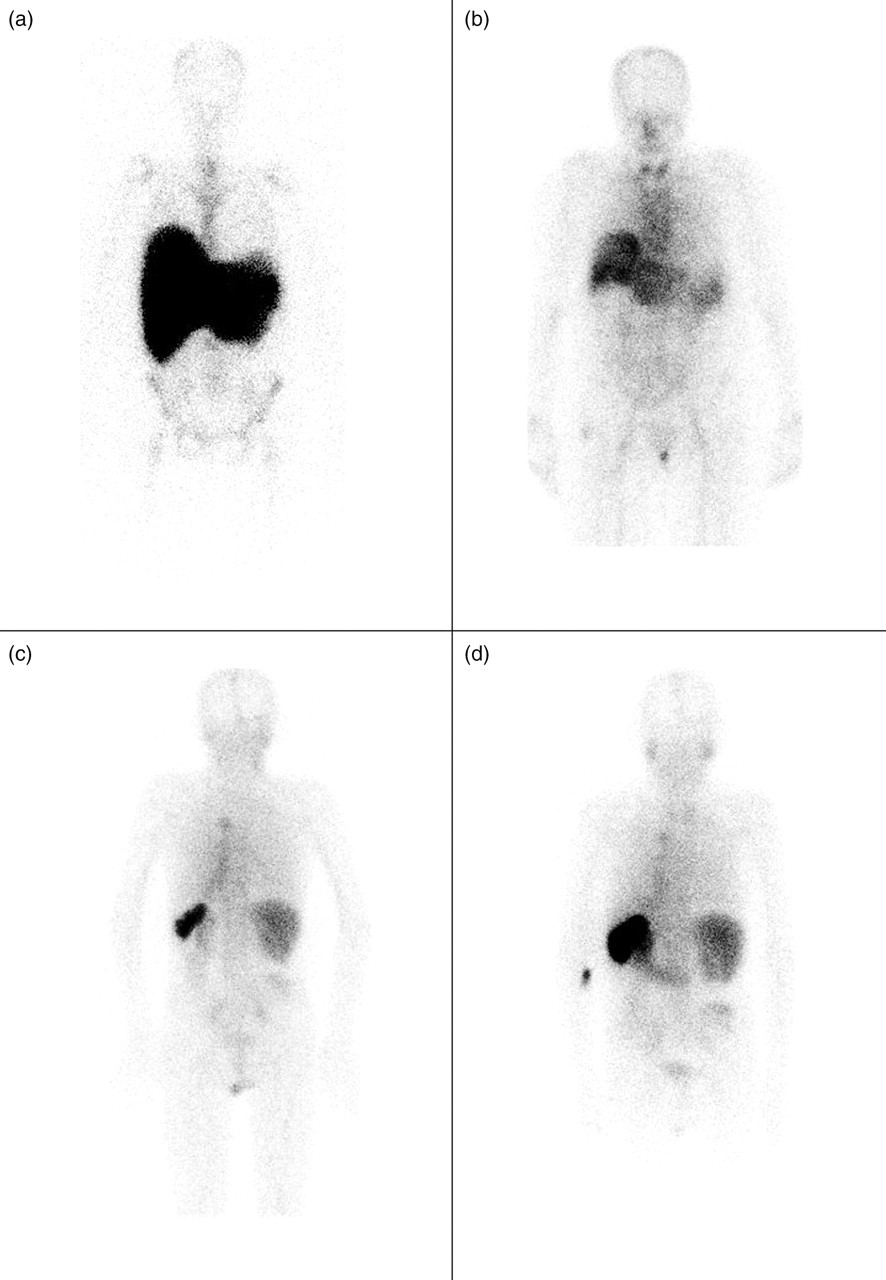
Since 5–10% of systemic amyloidosis is hereditary, genetic testing is often required, but the results must be interpreted in light of other findings, notably immunohistochemical or proteomic typing of the amyloid. 23–26 The clinical phenotype associated with particular mutations may vary, even within a given kindred, and since penetrance is variable, patients with AL amyloidosis can occasionally have an incidental mutation. 27,28 Conversely, some patients with hereditary amyloidosis have a potentially misleading but coincidental monoclonal gammopathy. 28,29
Biochemical analyses are an integral part of the diagnostic process. A plasma cell dyscrasia can be identified in approximately 94% of patients with AL amyloidosis. 30 Monoclonal proteins can be detected by serum and urine electrophoresis and immunofixation, although the fully quantitative high sensitivity serum free light chain (FLC) assay is usually best for serially monitoring progress and response to chemotherapy in AL amyloidosis. 31 Bone marrow examination and skeletal X-ray surveys are required to exclude frank multiple myeloma. It is, however, important to note that incidence of monoclonal gammopathy of undetermined significance occurs in at least 3% among people over 50 y, and demonstration of a plasma cell dyscrasia therefore does not by itself confirm amyloidosis is of AL type.
Diagnosis of amyloidosis is thus a multidisciplinary process, encompassing the clinical picture, various imaging modalities, histology, immunohistochemistry, proteomics, haematological and biochemical investigations and genetic analyses (Figure 4).
Diagnostic algorithm for the investigation of patients with suspected amyloidosis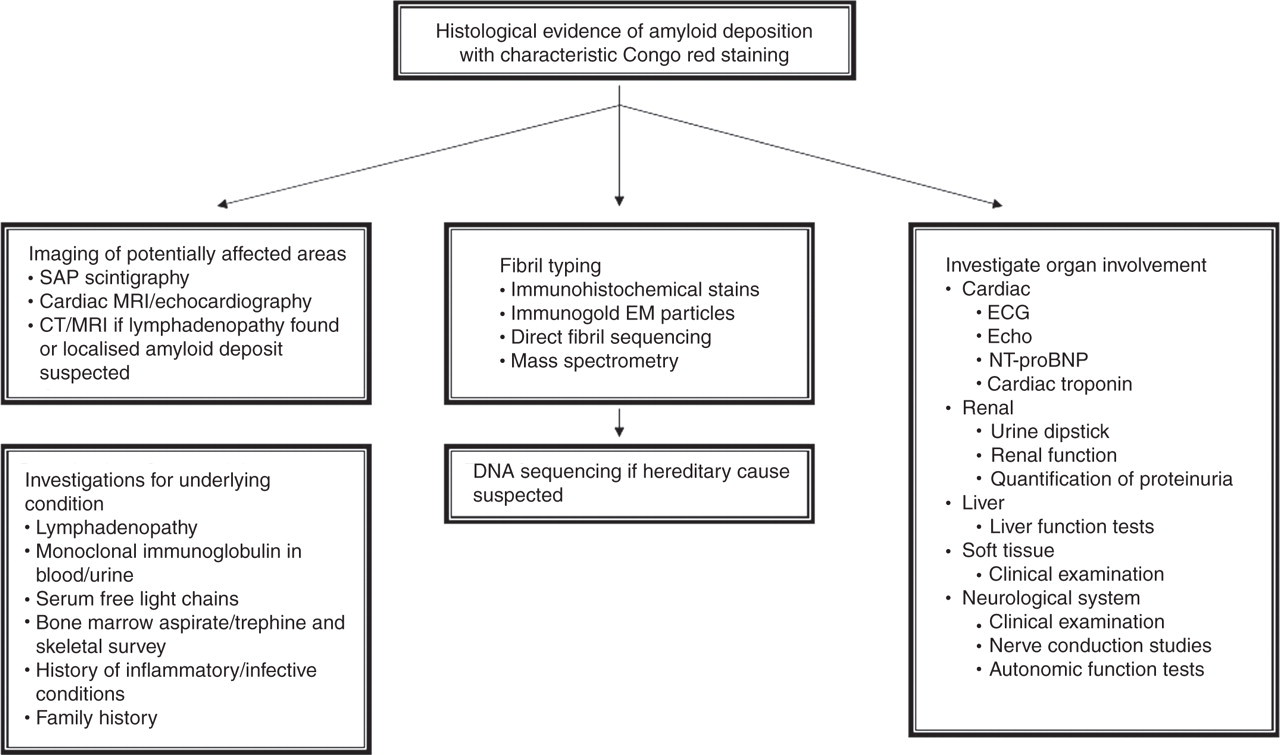
Localized amyloid
Localized amyloid deposition results from the local production of fibril precursor proteins. 32 Most clinically significant deposits are AL type, associated with foci of low-grade monoclonal B-cells which secrete monoclonal immunoglobulin light chains in the immediate vicinity. 33,34 The most frequent sites of deposition are the respiratory tract, the urogenital tract, the skin and orbits, but all are rare. 35 There are case reports of amyloid affecting almost any site ranging from intracranial amyloidomas, 36 amyloidoma affecting the larynx and oropharynx 37 to localized amyloid of the vagina. 38 Local resection of the ‘amyloidoma’ can sometimes be curative, 39 but amyloid deposits can recur within the same site or elsewhere in the same tissue. Amyloid deposits that appear to be localized can sometimes be a manifestation of systemic disease. It is therefore important to fully investigate patients in order to exclude systemic amyloidosis. 40 Once established that the amyloid deposit is localized, the management is dictated by the area involved and the degree of symptoms. Due to the extremely rare nature of the disease, management strategies have been somewhat experimental, ranging from radiotherapy, 41 to carbon dioxide laser ablation. 42 If the lesions are not causing symptoms, then clinical surveillance may well be all that is needed.
Localized masses of amyloid can be found at insulin injection sites when repeated administration to the same area has occurred over many years. 43 This form of amyloid stains with antibodies to insulin and has been termed iatrogenic A-Ins type amyloid.
Systemic amyloidosis
Four major acquired forms of systemic amyloidosis have so far been identified. These are systemic light chain amyloidosis (AL); systemic amyloid A amyloidosis (AA); dialysis-related amyloidosis (DRA); and senile systemic amyloidosis (ATTRwt).
Systemic AL amyloidosis
AL amyloidosis is the commonest form of amyloid and is thought to be the cause of death in 1/1500 people in the UK. The fibrils are formed of fragments of monoclonal immunoglobulin light chains consisting of all or part of the variable domain (VL).
44
AL amyloidosis is a rare complication of monoclonal gammopathies and can occur in association with any form of monoclonal B-cell dyscrasia.
30
The plasma cell proliferation fraction is usually similar to that of monoclonal gammopathy of undetermined significance (MGUS).
45
Approximately 15% of patients have multiple myeloma and within this group symptomatic myeloma is unusual.
30
Median age at presentation is 50–60 y and both sexes are equally affected.
30
Presentation is extremely variable as almost any organ can be affected except the brain. It is usually diagnosed incidentally on a biopsy. Clinical suspicion of amyloidosis should be raised in any patient with unexplained nephropathy, cardiac failure, peripheral or autonomic failure, hepatomegaly or splenomegaly or any unexplained multisystem disease. Approximately 50% of cases involve the kidneys presenting with proteinuria and frequently nephrotic syndrome. Kidney remains the commonest tissue from which the disease is identified. Patients with AL amyloidosis can develop acquired factor X deficiency, underlying some reluctance to perform biopsies in patients with AL amyloidosis. Factor X assays are important in patients with abnormal clotting, and replacement with fresh frozen plasma prior to biopsy in those who are deficient is recommended.
46
However, overall, the incidence of bleeding complications following renal biopsy in 138 patients with cast nephropathy or amyloid was no different from those without.
47
Cardiac involvement causing heart failure at presentation occurs in 15–30% of cases. Symptoms of cardiac involvement are those of congestive cardiac failure most commonly with progressive breathlessness.
30,48
Postural hypotension is common in patients with cardiac involvement. However, this can also be a feature of autonomic nerve involvement or intravascular depletion due to nephrotic syndrome. Liver involvement is the predominant presenting feature in relatively few cases but is a common finding on postmortem studies with between 62 and 90% reported.
49
Hepatomegaly and elevated alkaline phosphatase are the most frequent findings,
50
which do not always correlate with the amount of amyloid deposited. Hyperbilirubinaemia features late on in the disease process and is associated with a poor prognosis.
51
Soft tissue involvement is pathognomonic for AL amyloidosis; macroglossia is not seen in other forms of amyloid (Figure 5).
(a) Extensive peri-orbital bruising in a patient with AL amyloidosis. (b) Macroglossia with dental indentations in a patient with AL amyloidosis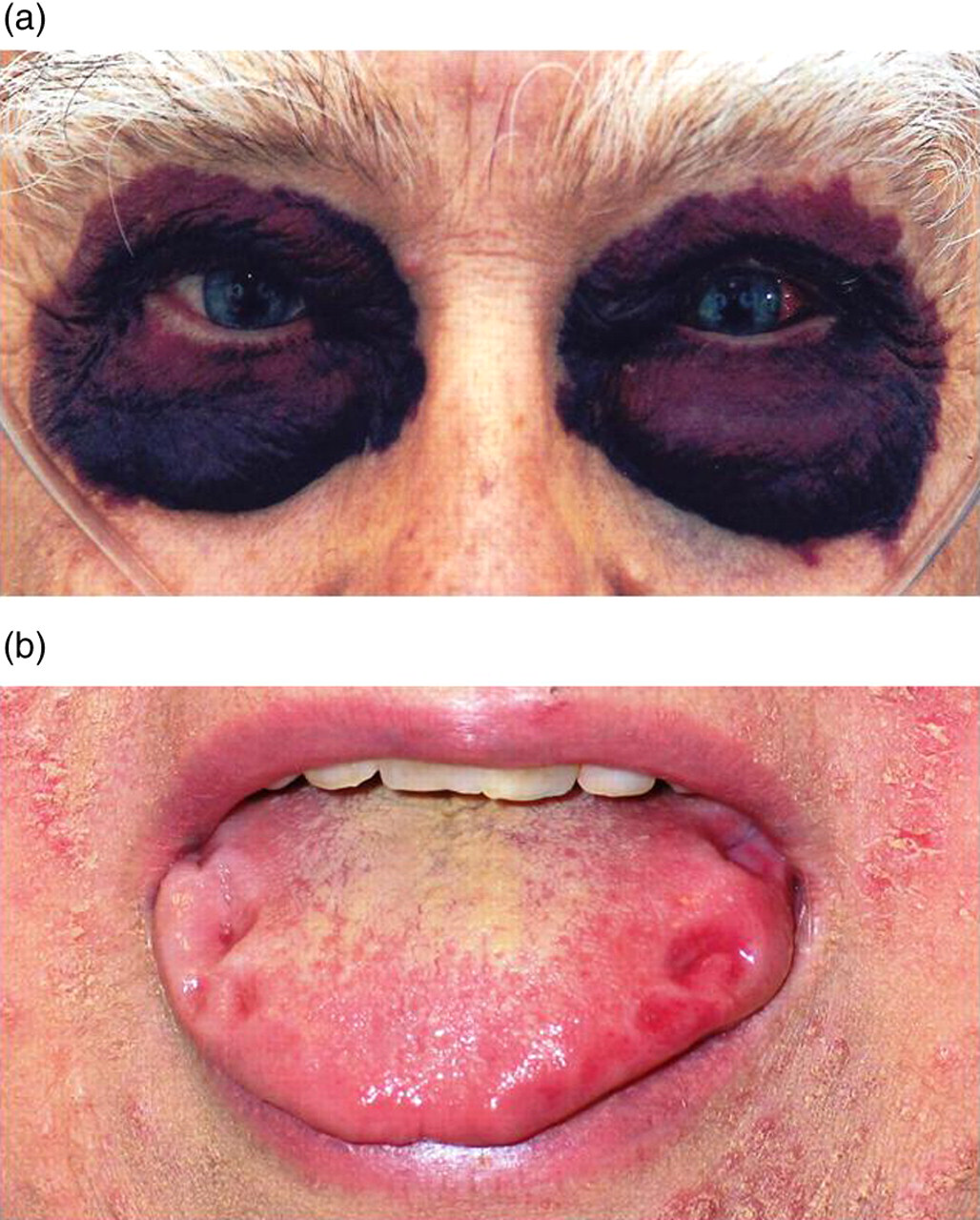
The prognosis in untreated AL amyloidosis is very poor with a median survival of only 6–15 months and a 10-year survival rate of <5%. 30 The prognosis, however, is somewhat dependent on the organs involved with cardiac and autonomic nerve involvement conferring a particularly poor prognosis. 30,48 In patients with renal involvement, factors associated with a poor prognosis at presentation are older age, elevated bilirubin and alkaline phosphatase, low serum albumin, low systolic blood pressure and higher serum free light chain concentration. 52 In 232 patients with cardiac involvement, patients with symptoms of congestive cardiac failure had shorter overall survival. 48 Echocardiographic features in patients with cardiac involvement such as shortened deceleration time and increased early diastolic filling velocity to atrial filling ratio have been shown to be predictors of death. 53
The current management of AL amyloidosis is to aim to suppress the production of the underlying B-cell clone with chemotherapy and in turn halt the production of amyloidotic light chains. Evidence suggests that remission of the underlying clonal disease is associated with preservation of organ function and in some cases remission of amyloid deposits. Early identification and treatment is associated with improved survival. 31 All current therapies in AL amyloidosis have been derived from experience in multiple myeloma, but adverse effects are far more frequent and severe in AL due to the multisystem nature of the disease. It is vitally important that an individual assessment is made for each patient and treatment regimens are tailored accordingly. A variety of therapies have been employed ranging from high-dose autologous stem cell transplantation to oral-based regimens. The mortality associated with autologous stem cell transplantation is significant. In the UK, over 10 y, the treatment-related mortality was 23%, which fell to 13% in the second half of the decade. 54 Higher mortality is associated with the number of organs involved, cardiac involvement, age, performance status and serum albumin level. 55,56 It is also important to note that stem cell transplantation itself poses a risk of end-stage renal failure, and in patients where preventing further renal decline is the goal of therapy, this should be carefully considered. Conventional non-transplant chemotherapy has been used for over 25 y in this disease and novel agents are constantly being trialled. Survival has significantly improved with the development of more therapeutic options. A recent report from the Mayo Clinic showed improved survival rates over time, with a most recent cohort from 2003 to 2006 having a 42% overall survival after four years. 57
Response to treatment is best monitored using serial serum free light chains in the majority of patients. The degree of haematological response needed to result in the gradual net regression of amyloid varies from patient to patient, reflecting the differing capacity among individuals to clear their amyloid deposits. Studies have shown that achieving a complete clonal response in patients who have received autologous stem cell transplants have better survival outcomes, 58 and patients who achieve a greater than 50% response to treatment were shown to have superior survival outcomes regardless of the treatment they received. 31 We have recently shown that in patients with AL amyloidosis and renal involvement, achieving a >90% dFLC (the difference between the involved and uninvolved light chain) response confers a survival benefit and also reduces the chance of renal progression, with a higher chance of achieving a renal response. 52 Whether the additional benefit of achieving a complete clonal response as compared with a >90% response outweighs the adverse effects of further chemotherapy necessary to achieve an improved response needs to be addressed in prospective studies.
Supportive management is imperative in this disease. Management of heart failure and nephrotic syndrome with diuretics is vital, especially during treatment when side-effects of agents such as dexamethasone and thalidomide can cause massive salt and water retention. Autonomic involvement is often extremely challenging to manage with hypotension and frequent collapses are a common occurrence in those severely affected. Fludrocortisone and midodrine can be helpful in some cases. Gastrointestinal (GI) symptoms secondary to direct gut wall infiltration or autonomic neuropathy can be particularly distressing for patients and difficult to manage. Patients frequently complain of bloating due to delayed gastric emptying and diarrhoea or constipation is common. Infection is a significant risk in all patients, especially in those with nephrotic syndrome, and patients should be vaccinated against seasonal flu. A low threshold for treatment with antibiotics should be employed. In patients with renal involvement, progressive renal decline is common. In our cohort of 752 patients with renal involvement and a baseline estimated glomerular filtration rate of >15 mL/min, 98 (13%) progressed to end-stage renal failure and required renal replacement therapy. 52 Outcome on dialysis was previously reported to be in the order of 12 months, but survival on dialysis is improving and in the UK, median survival on dialysis has now reached 39 months. 52
Transplantation in amyloidosis has been contentious due to concerns that patients may not survive long enough to benefit from transplantation and that amyloid may recur within the graft. We have reported the UK experience following solid organ transplantation and in fact very few patients develop clinically significant graft amyloidosis. Twenty-two patients had received renal transplants with a 67% five-year survival and no graft failures due to recurrent amyloid. Fourteen patients received cardiac transplants with a 45% five-year survival. In eight patients, cardiac transplantation was performed to enable a subsequent stem cell transplant and the median survival in this group was 9.7 y. 59 It is important to appreciate that these transplant recipients represent only 2% of all patients presenting with AL amyloidosis, i.e. a highly selected group.
Systemic AA amyloidosis
AA amyloidosis is a rare complication of chronic inflammatory conditions. The fibrils in AA amyloidosis are derived from the circulating acute phase reactant SAA, which is produced by hepatocytes under the transcriptional regulation of pro-inflammatory cytokines. 60 The median plasma concentration of SAA in health is <3 mg/L, but it can increase more than a thousand-fold during the acute phase response. 61 Longstanding elevation of SAA is a prerequisite to development of AA amyloidosis but it is rare and its incidence varies throughout the world, and for example seems less common in the USA compared with central Europe and Scandinavia. The commonest predisposing conditions in the Western world are the chronic inflammatory arthropathies, which account for over 50% of cases. In the developing world, reported cases are mainly associated with underlying infection. Patients with hereditary periodic fever syndromes are especially susceptible, perhaps due to the lifelong nature of these inflammatory diseases, and this risk is substantially increased when there is a family history of AA amyloidosis. Other rare causes include Castleman's disease, vasculitis and neoplasias such as lymphoma and mesothelioma. 62 Biopsy and postmortem studies have suggested a prevalence of up to 3–6% in rheumatoid arthritis (RA), 63,64 and 11–13% in familial Mediterranean fever despite availability of colchicine treatment for the latter. 65 The factors that govern susceptibility to AA amyloidosis in the face of a high SAA concentration are yet to be determined. Polymorphisms in the gene encoding for SAA1 isotype may contribute in part. 66
AA amyloidosis predominantly affects the kidneys, with more than 95% of patients presenting with proteinuria, and around 10% of patients having already reached end-stage renal failure at diagnosis. Splenic involvement is evident on SAP scintigraphy almost without exception, and deposits commonly occur in the adrenal gland, liver and GI tract, although usually without associated organ dysfunction. Cardiac and neuropathic involvements are extremely rare. 62
Patients with persistent inflammation frequently develop progressive renal dysfunction and end-stage renal failure within 5–10 y. Almost 60% have nephrotic syndrome at presentation, which confers a high risk of infection. Acute kidney injury is common and is often non-reversible, emphasizing the need for great care with regard to hypoperfusion, nephrotoxic drugs and surgery. Patient outcome has gradually improved, with a recently reported median survival of 133 months. 62 Outcome is poorer in association with older age, lower serum albumin and end-stage renal failure at presentation. Serum SAA concentration has a powerful and modifiable influence on outcome; complete suppression of inflammation in terms of SAA concentration persistently <5 mg/L is frequently associated with regression of amyloid and preservation of renal function. Treatment will differ according to the nature of the underlying chronic inflammatory disorder, and there has lately been much progress with biological therapies for rheumatoid arthritis, etc., but progressive renal dysfunction remains common and end-stage renal failure occurs in up to a third of patients. Median survival on dialysis is in the order of 4–5 y, which is similar to that among age-matched non-diabetic patients; renal transplantation has been performed in selected cases with reportedly excellent outcomes.
Dialysis-related amyloidosis
DRA is a complication of long-term dialysis following end-stage renal failure. The underlying fibril is due to β 2-microglobulin (β 2M). β 2M is the light chain component of the major histocompatibility complex (MHC) class 1 molecule. It is synthesized in all cells that express MHC class 1 molecules. 67 β 2M is cleared from the body by the kidney. It is freely filtered by the glomerulus and reabsorbed by the proximal tubular cells. 68 When patients develop end-stage renal failure, β 2M accumulates and the circulating concentration rises from normal levels (1–2 mg/L) to 50–70 mg/L. Most cases present clinically after 10 y on dialysis and this form of amyloid has a tropism for osseo-articular surfaces. Symptoms of DRA manifest as carpal tunnel syndrome, arthralgia, spondyloarthopathy, subchondral bone cysts and fractures. While modern high flux dialysis techniques may have reduced the incidence of β 2M amyloidosis, renal transplantation remains the only effective treatment in reducing the symptoms of established diseases. 69
Senile systemic amyloidosis
ATTRwt is a disease of the elderly and usually affects men; the fibril is composed of normal wild-type transthyretin. 70 Amyloid deposits of this type are found at autopsy in about 25% of patients over age 80, with predominant involvement of the heart. 71 Non-clinically significant deposits are frequently present in other sites including the lungs, gut, bladder and small arteries in many other tissues. 72 A relatively small proportion of patients with wild-type transthyretin amyloid deposits present with clinical disease, and among those who do, most present with restrictive cardiomyopathy and congestive cardiac failure. Wild-type transthyretin-derived amyloid has been found in specimens following carpal tunnel release surgery, 73 which often precedes cardiac manifestations. 74 Echocardiography demonstrates a markedly thickened myocardium, which is typically greater than in AL amyloidosis. In a study of 18 patients with ATTRwt, the mean interventricular septum thickness was 17.8 mm in the ATTRwt group as compared with 14.3 mm in the AL group (P = 0.002). 75 Identification of ATTRwt has lately increased due to the advent of cardiac magnetic resonance imaging, but definitive diagnosis continues to rest on endomyocardial biopsy confirming amyloid of transthyretin type in conjunction with wild-type TTR gene sequence. 76 The natural history is slow progression of heart failure with much better median survival than AL amyloidosis at 75 months in one recent series. 75 The mainstay of management is supportive care and symptomatic management of heart failure with fluid restriction, low-salt diet and diuretics.
The hereditary systemic amyloidoses
Hereditary amyloidosis is a group of diseases due to mutations in different specific proteins. All are autosomal dominant but penetrance is variable and there is often no family history. Age of onset, disease penetrance and phenotype vary widely between different mutations and even within kindreds, presenting a challenge for genetic counselling. Hereditary systemic amyloidosis can be divided into neuropathic and non-neuropathic forms. The former comprise familial amyloid polyneuropathy (FAP), usually caused by mutations in the transthyretin gene (ATTR) and gelsolin amyloidosis (AGel). Non-neuropathic forms include fibrinogen Aα-chain amyloidosis (AFib), apolipoprotein AI amyloidosis (AApoAI), apolipoprotein AII amyloidosis (A ApoAII) and lysozyme amyloidosis (ALys).
Hereditary transthyretin amyloidosis (ATTR, familial amyloid polyneuropathy)
The most common type of hereditary amyloidosis worldwide is associated with mutations in the gene for transthyretin. There are some 100 point mutations associated with the clinical syndrome, which is typically characterized by progressive peripheral and autonomic neuropathy and is known as familial amyloid polyneuropathy (FAP). Cardiac amyloidosis is extremely frequent, as are varying degrees of amyloid deposition in the viscera, vitreous humour, gut and occasionally the central nervous system. 77,78 Clinical presentation is typically from the third decade, although this varies with mutation. 79,80
Transthyretin is a transport protein that circulates in a tetrameric form. More than 95% of transthyretin is produced in the liver, with the remainder in the choroid plexus and in the retina. 81,82 The most common transthyretin mutation involves the substitution of methionine for valine at position 30 (ATTRV30M). This usually presents with sensori-motor peripheral neuropathy and, unlike most other mutations, cardiac involvement is rare. The disease typically develops by age 30–40 y in the Portuguese focus, but about 20 y later in the Swedish one. The most common aetiology of FAP in the UK and Ireland is the T60A variant. 83 This usually presents after the age of 50 and often with autonomic symptoms, but cardiac amyloid is virtually always present at diagnosis. 84 Three to four percent of black individuals have the V122I transthyretin variant, which is associated with a predominantly cardiac phenotype that is clinically indistinguishable from senile cardiac amyloidosis; it usually presents after age 60 and is not associated with neuropathy. 85
Familial amyloid polyneuropathy is a fatal progressive disease with a life expectancy of around 10 y after symptoms have developed. Individuals with early-onset disease may experience more rapid progression, whereas progression is reportedly slower and with survival of up to 20 y among patients who present after age 55–60 y. 86
TTR amyloidosis has become a particular focus for development of novel anti-amyloid therapies, both with a view to stabilizing soluble TTR in the blood and inhibiting its production through silencing RNA and antisense oligonucleotide approaches, discussed below. Several such strategies have already progressed to clinical trial. However, the only therapy that has been adopted into widespread clinical practice is orthotopic liver transplantation (OLT), which was introduced in 1990 on the basis that almost all mutant TTR is produced in the liver. 87,88 Since this date, over 700 transplants have been performed. The Familial Amyloid Polyneuropathy World Transplant Registry (FAPWTR) reported seven years of registry data in 2003. The most common mutation was ATTRV30M, representing 83% of cases, and five-year survival was significantly better in this group when compared with patients with non-ATTRV 30M mutations (79% versus 56%, respectively, P < 0.001). Symptoms of peripheral neuropathy have been reported to gradually improve in up to 50% of cases, 89 and better outcomes have been associated when transplantation has been performed earlier on in the course of the clinical disease. 84 However, OLT remains controversial. Cardiovascular death is higher in patients with FAP as compared with other indications for liver transplantation and progression of cardiac amyloidosis can occur afterwards due to ongoing deposition of wild-type TTR in this particular anatomical site. 90,91 The indications, timing and outcome of liver transplantation for FAP thus remain unclear, as indeed does its role in patients with non-ATTRV30M mutations who already have cardiac amyloid deposits.
Hereditary gelsolin amyloidosis
Hereditary gelsolin amyloidosis is also known as amyloidosis of the Finnish type. The amyloid fibrils are derived from cleavage fragments of variant gelsolin. It was first described by the Finnish ophthalmologist Jouko Meretoja in 1969. 92 This disease has since been reported in various other countries but the largest population remains in Finland. Two variants have been found, G654A and G654T, and both have been reported in Finland. The G654A type has also been reported in Portugal, Japan and Iran whereas patients with the G654T variant have been reported in Denmark, the Czech Republic and France. 93 Gelsolin is an actin-modulating protein that enhances migration of cells. Mutated gelsolin is unable to bind calcium ions and it is thought that this may render it more prone to proteolysis and subsequent fibril formation. 94–96
The disease typically presents with corneal lattice dystrophy during early middle-age and develops as a slowly progressive but very disabling and deforming cranial neuropathy. 97 Life expectancy is near normal, and despite substantial renal amyloid deposits being present at an early stage, there is usually no associated clinical evidence of visceral involvement. Rare cases of homozygous mutations have been reported with a rapid decline and renal failure 98 and renal dysfunction can occur in heterozygotes.
Fibrinogen Aα-chain amyloidosis
Hereditary fibrinogen amyloidosis (AFib) was first characterized in a Peruvian kindred in 1993 99 and it is the most common cause of hereditary renal amyloidosis in the UK. However, disease penetrance is variable and a family history is very often absent. To date, nine amyloidogenic mutations in fibrinogen have been identified with the E526V variant being much the commonest. Presentation is universally with proteinuria and renal impairment and the diagnosis is almost invariably made on renal biopsy. Histological findings show a characteristic picture of massive glomerular amyloid infiltration with almost complete obliteration of the normal architecture but little or no vascular or interstitial deposits. Hypertension is common. Extra-renal amyloid deposits are usually evident in the spleen and sometimes adrenal glands on radiolabelled 123I-labelled SAP scintigraphy; liver involvement is rare, but can be clinically significant. 24 Cardiac involvement has been reported in a few non-E526V-associated cases. Presentation typically occurs around age 60 y, with progression to end-stage renal failure within about five years from diagnosis. The absence of clinically significant extra-renal disease is consistent with median survival from presentation of 15 y. 24 Median graft survival following isolated renal transplantation is in the order of seven years, with recurrent amyloid disease contributing to graft loss beyond this stage. On the basis that the amyloidogenic protein is produced exclusively by the liver, combined liver and renal transplantation has been offered to some younger patients who have developed end-stage renal disease, with an excellent outcome in about half of cases. While further amyloid deposition is prevented by these means, there has been substantial early mortality; three out of nine reported cases having died due to complications of surgery. 100
Hereditary apolipoprotein AI amyloidosis
Apolipoprotein AI is produced in the liver and intestines, and is catabolized in the liver and kidneys. It is the predominant protein in high-density lipoprotein 101 and plays a key role in reverse cholesterol transport. 102 While tiny wild-type apolipoprotein AI-derived (AApoAI) amyloid deposits have been identified in atherosclerotic plaques, some 13 variants have been associated with major visceral amyloidosis, i.e. hereditary systemic amyloidosis. The pathogenesis involves proteolytic cleavage, with the amino terminal 83–93 residues typically being incorporated into the amyloid fibrils. 86 Currently, of the more than 50 ApoAI variants known, 13 are associated with amyloidosis. 103 Different mutations are associated with a variety of phenotypes and again there is marked variability even within families which makes genetic counselling challenging. Chronic renal failure is the most common manifestation but there may also be significant neurological, cardiac and hepatic dysfunction. The phenotype of the following six variants (Gly26Arg, Trp50Arg, Leu60Arg, Del70–72, Leu75Pro and Leu64Pro) is characterized by renal manifestations in association with extensive visceral amyloid deposits and hepatosplenomegaly. Clinical presentation is typically with hypertension and proteinuria between 18 and 55 y of age. Patients with the Leu75Pro variant have reportedly presented as late as their seventh decade and survived into their 90s. 29,104 Progression of renal impairment is slow with a median time to end-stage renal failure from presentation of eight years in one series. A large load in the liver frequently occurs in association with elevated serum alkaline phosphatase and gamma-glutamyl transpeptidase, but synthetic function is rarely affected and usually is a late feature after many years of progressive amyloid deposition and fulminant hepatic failure is extremely rare. 105 Several ApoAI variants (Leu90Pro, Arg173Pro, Leu174Ser and Leu178His) are associated with skin and cardiac amyloid deposits with death usually occurring due to progressive cardiomyopathy within 10 y of diagnosis 106–109
It is thought that at least 50% of ApoAI is produced in the liver. 110 Liver transplantation has been associated with regression of extra-hepatic amyloid in a number of cases, 111 the reduction in supply of variant ApoAI by around 50% evidently being sufficient to alter the balance of amyloid deposition and its natural turnover in favour of the latter. ApoAI amyloidosis is a slowly progressive disorder and reports following renal transplantation have shown remarkable graft survival, frequently exceeding 10–15 y despite histological evidence of recurrent amyloid in the transplanted organ. 110 Liver transplantation has been performed very rarely, but would appear to have the potential to benefit patients with extra-renal amyloidosis, notably peripheral nerve and cardiac involvement, or of course those with progressive liver dysfunction.
Apolipoprotein AII amyloidosis
Apolipoprotein AII amyloidosis was first described by Weiss and Page in 1973. 112 It is the second most abundant HDL apolipoprotein. Four amyloidogenic mutations have been reported to date. Presentation is with proteinuria and progressive renal impairment. Age of onset tends to be earlier than for AApoAI with cases requiring renal replacement therapy by age 30–40 y. Russian and Spanish kindreds have reported later onset with presentation age 30 y and onset of renal replacement therapy at age 50 with the Stop78Arg mutation. 113 Outcome following transplantation has been reported to be excellent but the number of reported patients is very small.
Lysozyme amyloidosis
Lysozyme amyloidosis was first described by Pepys et al. in 1993. Lysozyme is a bacteriolytic enzyme found in high concentrations in the liver, articular surfaces, saliva and tears. It is highly expressed in granuloctes, monocytes and bone marrow precursor cells. To date, seven amyloidogenic mutations have been found: Ile56Thr, Phe57Ile, Trp64Arg, Asp67His, Trp112Arg, Tyr54Asn and Asp67Gly. Clinical presentation is with very slowly progressive renal failure, usually in the third and fourth decades. Involvement of the liver, lymph nodes, GI tract and spleen also occurs. Sicca syndrome due to salivary amyloid deposition is frequent in patients with Try64Arg and Asp67His variants, and lung and thyroid deposits have been reported in patients with Ile56Thr. 114 Renal decline can take decades, and the few reports of renal transplantation have been excellent, with reported graft function of over 15 y in some patients.
Therapeutic targets
To date, the objectives of therapy in amyloidosis have comprised supportive measures including renal replacement therapy and occasionally organ transplantation, coupled with efforts to suppress production of the respective amyloidogenic precursor protein, for example through chemotherapy in AL type. The latter is frequently associated with gradual regression of existing amyloid deposits and preservation of vital organ function, and associated improvement in survival. 31,115 Although great strides have been made in the management of AL and AA amyloidosis, this strategy still often fails and it has not been applicable in many other types of amyloid. This has led to the search for new drug therapies that either inhibit amyloid deposition or enhance removal of established amyloid deposits.
TTR amyloidosis has emerged as a particular focus for novel drug development, and several novel approaches are being pursued. The native soluble TTR molecule is a tetramer comprising two dimers that create and span a thyroid hormone-binding pocket. It is thought that a requisite step in the transformation of soluble TTR to its amyloid form is disruption of the normal TTR homotetramer into its monomeric components, which can auto-aggregate in the misfolded but highly ordered fibrillar conformation. 116 The approach pursued by Kelly and his colleagues has been to identify ligands that occupy the thyroid hormone-binding pocket and stabilize the structure in its native tetrameric form. 117 This has led to the development of a novel drug compound tafamidis, which has shown promise in a randomized trial in hereditary TTR amyloidosis associated with the Met30 variant (V30M). 118 Our group at UCL has identified other very potent stabilizing compounds that simultaneously occupy both T4 binding sites in each tetrameric TTR molecule, confirmed by X-ray crystallographic analysis, which is irreversible under physiological conditions, and inhibited amyloidogenic aggregation more potently than other known ligands. 116 The hepatic origin of circulating TTR has encouraged development of RNA inhibiting approaches to treatment, given the preferential potential of targeting the liver with these new technologies. Both silencing RNA and antisense oligonucleotide approaches, which aim to reduce production of TTR, are being developed for clinical trials.
A completely novel therapeutic approach has been to directly target the amyloid deposits. SAP is present in all amyloid deposits and its presence may mask their recognition and efficient clearance by natural systems that evidently do operate at low levels. 119 Further, amyloid formation is inhibited in SAP knockout mice. 120 The precise role of SAP in fibrillogenesis remains unclear but Pepys identified its role as a potential therapeutic target in 1984, 121 which led to the development of (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexa-noyl]pyrrolidine-2 carboxylic acid (CPHPC). This palindromic bis(D-proline) compound cross-links SAP in the plasma triggering its clearance by the liver, resulting in almost complete depletion of circulating SAP. 122,123 A preliminary clinical study of CPHPC in several forms of amyloidosis confirmed that regular administration produced sustained and profound depletion of SAP. The drug was given to several patients for many years with no apparent adverse effects, although the magnitude of potential clinical benefit was not sufficiently large to be ascertained in this open, non-controlled study. 124 Most recently, the observation that CPHPC efficiently depletes SAP from the blood, but only very slowly from the amyloid deposits, has enabled development of passive immunotherapy targeting the residual amyloid-associated SAP. This approach results in rapid and very substantial clearance of experimentally induced amyloid deposits by macrophages, and is currently in translation for patients. 125
Conclusion
Amyloidosis is a diverse group of multisystem diseases in which much progress has been made in recent years. There have been major developments in diagnosis, ranging from scintigraphic imaging methods and cardiac MRI, to improved molecular characterization of amyloid deposits through mass spectrometry and genetic sequencing. Clinical outcomes have improved through advances in chemotherapy, biologic anti-inflammatory agents and organ transplantation in AL, AA and hereditary amyloidosis, respectively. Assessment of underlying diseases and amyloidotic organ function with biochemical markers such as the serum free light chain assay, SAA measurements and the cardiac biomarker NT-proBNP, have greatly improved the monitoring of patients. A new era of clinical trials to test the first drugs specifically designed to prevent or remove amyloid deposits is upon us, promising a brighter future for patients with these very serious diseases.
DECLARATIONS