
Abstract
Neurological dysfunction results from vascular, inflammatory, degenerative, neoplastic, metabolic or genetic causes. Of particular interest is a group of neurological symptoms thought to be linked to an underlying tumour, the so-called paraneoplastic syndromes. It is considered to be due to an attempt by the immune system to subjugate the growth of the tumour by triggering an antibody response against the neuronal antigens expressed by the neoplasm. The unfortunate consequence of this is an assault by the immune components on the nervous tissue, thereby rapidly precipitating a variety of neurological deficits. Every level of the nervous system is potentially vulnerable, with the disability being considered as irreversible due to the lack of regenerative capacity of the neurons. This phenomenon is rare, occurring at an approximate frequency of less than 1% of all tumours and often accompanied by the presence of specific high-titre autoantibodies in both the cerebrospinal fluid and blood. This group of antibodies are non-pathogenic markers for paraneoplastic neurological syndromes, which have expanded to almost 20 since the discovery, in 1986, of the first clinically relevant syndrome. More recently, a new generation of antineuronal antibodies against cell surface antigens, having a direct pathogenic role in causing the disease, has emerged to complement the existing repertoire. Neuronal antibodies are useful diagnostic markers of the brain disease and also, in some cases, may reveal an underlying malignancy, thus facilitating faster diagnosis and earlier treatment with consequently better prognosis.
Introduction
For the past five decades, the detection and identification of autoantibodies have been very useful in the assessment of systemic and organ-specific autoimmune diseases. Currently, more than 100 clinically relevant autoantibodies have been documented in human autoimmune disease, including those associated with the disorders of the nervous system.
1
The underlying autoimmune processes are widely thought to involve:
A non-specific inflammatory response (known as innate immunity – a first line of defence usually against an invading pathogen or non-infectious agent, relying on polymorphonuclear cells, monocytes, natural killer cells and biologically active peptides); An adaptive immune response (comprising of a specific immune reaction of the lymphocytes and antibodies against the pathogen/antigen).
These act as major components in eliminating any agents (e.g. cancer) that might compromise health.
Among the markers of the autoimmune disease, some are credited with causing the disease (for example acetylcholine receptor [AChR] antibody in myasthenia gravis) while many others are envisaged as merely an epiphenomenon of the disorder (for example, oligoclonal bands in multiple sclerosis [MS]; Table 1). A further inclusion in the latter category is a newer generation of rare immune indicators for paraneoplastic nervous system diseases. These disorders have been generally difficult to diagnose universally, as the knowledge and experience have been confined to the specialist centres. This is further compounded by the low number of cases and lack of positive control materials available to the non-specialist centres. Besides the confirmation of autoimmunity to nervous tissue, these immune markers may indicate the presence of an underlying malignancy. Recently, several antibodies have been described which may or may not be associated with malignancies, but have well-defined clinical syndromes associated with them. The focus of this paper will be confined to specific antibodies associated with complex neurological manifestations, with or without an underlying malignancy. The significance of other immune markers of neurological diseases (e.g. oligoclonal bands in MS) will not be discussed further.
Disorders of the nervous system and their markers
MS, multiple sclerosis; NMO, neuromyelitis optica; GBS, Gullain-Barré syndrome; MG, myasthenia gravis; AChR, acetylcholine receptor; VGKC, voltage-gated potassium channel; VGCC, voltage-gated calcium channel; GAD, glutamic acid decarboxylase; NMDAR, N-methyl-
*The exact target for VGKC antibodies have recently been identified to be some of the complexing proteins associated with the potassium channels (e.g. CASPR2, LGI1, etc.)
Antibodies in italics have a confirmed pathogenic role
Historical background
In the last two decades, scientific advances have furnished clinical immunology laboratories with tests for a number of rare autoimmune diseases such as paraneoplastic neurological syndromes (PNS). Historically, such neurological disorders were first linked to remote cancers in the 19th century by Auchè, 2 a French physician. Much later, in the mid-20th century, a surge in the clinical interest in this area led to the generic term ‘paraneoplastic neurological syndromes’ (PNS) being coined to encompass any neurological deficit associated with a neoplasm, but is not caused by the tumour or its metastases and occurring in the absence of any other known cause. 3 The designated expression, PNS, is usually reserved for a rare group of disorders that have immune-mediated pathogenesis, a concept proposed in 1951 based on circumstantial evidence. 4 Only recently, concrete support for immune-mediated involvement in the pathogenesis was provided by Posner's team: serum antineuronal nuclear antibody (ANNA-1) was found in a patient with neurological syndrome and cancer. 5 This antibody reacted specifically with the patient's tumour and also neuronal tissue, suggesting that similar antigens were expressed in both tissues, thus supporting the idea of natural immunity to cancers. 6,7 Posner's observations 5 first availed the laboratories with a new diagnostic test for an important but rare neurological condition. With the advent of this observation, currently, there exists a range of antineuronal antibody markers, known as paraneoplastic neurological antibodies (PNAs, also known as onconeuronal antibodies), which complement the diagnosis of these clinical syndromes. These markers are organized into two broad categories that determine whether these specificities are pathogenic or not (Figure 1). The pathogenic nature of an antibody appears to be governed by the location of its antigen and is synonymous with its binding to an extracellular site, while the non-pathogenic antibodies would have intracellular antigenic targets. These can further be subcategorized into paraneoplastic if there is a clear association of the antibody with the cancer (which can sometimes be identified several months after the neurological symptoms), or non-paraneoplastic when a cancer is not identified after extensive investigations and over time. 8 Prior to embarking on the autoimmune markers, it would be pertinent to pay due consideration to the affiliated clinical diagnoses.
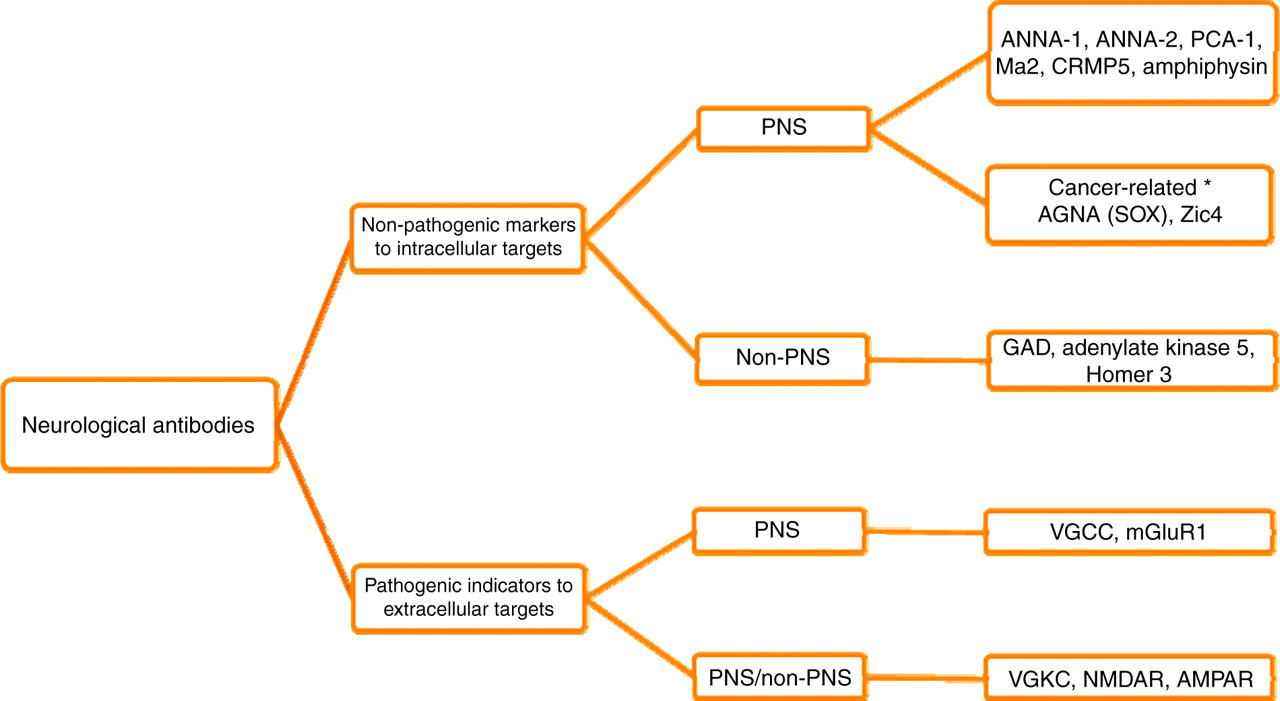
Classification of neurological antibodies. *Although cancer-specific antibodies but pathology not immune-related.
Clinical presentations
PNS may initially present with a variety of neurological symptoms ranging from tingling/numbness in the extremities, difficulty in walking, slurred speech, confusion, hallucinations and memory impairment to seizures. Other non-specific symptoms like unexpected weight loss, night sweats, fever or myalgia may suggest a neoplastic process in some patients.
PNS that almost always accompany a cancer are known as ‘classical’ syndromes. 9 Identifying a ‘classical’ neurological disorder (Table 2) in conjunction with the paraneoplastic antibodies can lead to a definitive diagnose of PNS and is an early indication of an underlying malignancy. These are briefly described below.
A summary of pathological findings in areas of the brain leading to the signs and symptoms seen in paraneoplastic neurological syndrome
Encephalomyelitis is due to inflammation of the brain and spinal cord and presents with a variety of symptoms including weakness, numbness, breathing difficulties, confusion and seizures.
Cerebellar degeneration, as the name suggests, is related to the cerebellar damage, therefore influencing control of muscle function and balance. Malfunction of the cerebellum can frequently lead to wide-based unsteady gait (ataxia) with limb or body tremors. Patients usually have difficulty co-ordinating their upper limb movements and have oscillating eye movements (nystagmus).
Limbic or brainstem encephalitis involves the inflammation of the limbic system and/or brain stem, resulting in memory loss, drowsiness, confusion, disorientation and seizures. Some of these syndromes may be subacute (happening over several days), but occasionally they can be chronic (over several weeks/months).
Opsoclonus-myoclonus results from damage to the cerebellum causing rapid, irregular eye movements (opsoclonus) accompanied with quick involuntary muscle jerks, especially of the limbs (myoclonus).
Lambert-Eaton myasthenic syndrome (LEMS) is due to disruption in the propagation of nerve impulses at the neuromuscular junction with consequent muscle weakness, fatigue, swallowing difficulties, dry mouth or blood pressure changes.
Neuropathies are caused by damage to the peripheral nerve predominantly affecting the autonomic or sensory pathways. Autonomic neuropathy presents as lack of involuntary control (variability in heart rate, blood pressure, etc.), changes in sweating, dry mouth, bladder dysfunction or impotence. Sensory neuropathy can manifest as numbness, pins and needles, tingling, pain or itching.
In addition to the above, other neurological disorders that can sometimes be seen with malignancies (‘non-classical’ syndromes) can be associated with well-defined antibodies. These include stiff-person syndrome (SPS; progressive, severe muscle rigidity affecting the spine and legs which may be accompanied by painful muscle spasms), myasthenia gravis (weakness and rapid fatigue of voluntary muscle-related activities such as facial and eye movement, chewing/swallowing, talking/breathing and arms/legs) and neuromyotonia (abnormal motor nerve impulse causing twitching, progressive stiffness, muscle cramps and sometimes muscle weakness).
Pathogenesis
The pathophysiology of PNS is complex and can affect any level of the central or peripheral nervous system. The precise mechanisms involved are unclear; however, it is generally understood to be due to neuronal antigens expressed in the tumour cells that elicit an immune reaction which is misdirected against common epitopes found in the neurons. This process can be considered as ‘side-effects’ of the immune system, attempting to curtail the tumour growth through the involvement of other components of the immune system, particularly the dysfunction of T-regulatory cells. 10 In addition, tumours themselves produce and release bioactive substances such as hormones, precursors, enzymes and/or cytokines which also contribute to inflammatory damage. This complexity and the involvement of multiple antigenic targets may result in the wide variation of the symptoms seen in these patients.
In the non-paraneoplastic syndromes, especially those associated with neuronal surface antibodies, the antibodies are likely to be directly pathogenic, although the exact trigger is not clearly known. A summary of the pathology and immune-mediated pathogenesis in some of these syndromes is given in Table 2.
Tumours associated with paraneoplastic syndromes
The tumours associated with PNS are histologically identical to any other non-PNS-related tumour, except for the infiltration of immune mediators such as T-cells, which may be responsible for their relatively smaller size and hence the difficulties in detecting them. 11 These tumours are best detected using a highly sensitive 18-fluoro-deoxyglucose positron emission tomography (FDG-PET) scan, especially when the initial screening by a computed tomography scan appears negative. It appears that these tumours are less aggressive and respond better to chemotherapy than those not linked to PNS 12 and therefore result in an improved survival time.
Animal model of the disease
In order to establish animal models of PNS, attempts to reproduce clinical signs/symptoms and induce pathological changes in rodents have been made. These have been generally unsuccessful, irrespective of the techniques used (immunizing with intracellular recombinant antigens, passively transferring circulating paraneoplastic antibodies with and without complement or injecting activated patient's lymphocytes 13 ). The results of these experiments support the concept that PNAs do not have a direct pathogenic role in the pathophysiology and that alternative immunological mechanisms are involved in the accompanying neuronal damage. The non-pathogenicity of these antibodies may be due to the viability of the neurons which restrict antibody access to intracellular antigens. It should be mentioned that rats injected with IgG from a patient with antiamphiphysin antibodies demonstrated dose-dependent stiffness and spasms. 14 However, it cannot be proven whether the pathogenic antibody is that which is directed against amphiphysin or another, as yet unidentified component in the IgG fraction. On the other hand, passive transfer animal models have been established in many of the neurological diseases like myasthenia, LEMS or neuromyelitis optica which are associated with pathogenic antibodies against cell surface antigens. 15–17
Methods of antibody detection
Tissue specific (immunocytochemistry)
Antineuronal antibodies tend to be present in high titres and are very often identified on cerebellar tissue from primate or rodents. Our approach is to define antibodies by the immunocytochemical distribution pattern seen during screening on primate cerebellum using indirect immunofluorescence. One advantage of this method is that in addition to the full range of characterized antibodies, other specificities can also be determined (Table 3). The usage of live unfixed neuronal cultures may enhance the sensitivity of this technique, but is more difficult to perform on a routine basis.
Range of antibodies that can be detected on primate cerebellum and their cellular location
Alternative names are given in the brackets
Please see Table 1 for definitions of abbreviations used in this table
Any positive serum sample identified by immunocytochemistry is subjected to further testing to confirm the specificity of each antibody using an alternative method such as commercial line blot painted with several recombinant proteins. The latter avenue confers additional advantages, particularly where there are coexisting PNAs: for example, antineuronal nuclear antibody type 1 (ANNA-1) occurring concurrently with collapsin response mediator protein type 5 (CRMP5) 18 or antineuronal nuclear antibody type 2 (ANNA-2) 19 or when antinuclear antibody is present. On occasions, tissue-specific antibodies like mitochondrial are present in sufficient concentration to obscure the identification of neuronal-specific antibodies. 20 Enzyme-linked immunosorbent assays (ELISAs) can also be utilized for confirmation purposes, but this is not versatile and may not be as economical as blots painted with several antigens.
Among six most commonly tested antibodies, Purkinje cell antibody (PCA-1), ANNA-1 and ANNA-2 are the easiest to recognize immunocytochemically as long as the non-neuronal cross-reactivities are taken into consideration. With appropriate experience, CRMP5, paraneoplastic neuronal antigen Ma2 (Ma2) and amphiphysin staining patterns can also prove to be fairly easy to recognize during routine testing.
Cell-based assays
Brain tissue lacks sensitivity in detecting certain antibodies, particularly those associated with extracellular antigens and to overcome this shortfall, molecular techniques have been utilized to design sensitive cell-based assays to provide an alternative substrate. Briefly, cDNA for various antigens (e.g. N-methyl-
Immunoprecipitation techniques
This is a quantitative technique that entails precipitating an antigen out of solution with a specific antibody that has high affinity for that particular antigen. In the case of voltage-gated calcium channels (VGCC), a radiolabelled synthetic peptide, ω-conotoxin MVIIC combines with the solubilized P/Q subunits, indirectly radiolabelling the VGCC. This, when incubated with the test serum, forms a complex with the anti-VGCC IgG. The radioactivity measured in the precipitated complex is proportional to the concentration of the antibody. 22
Voltage-gated potassium channel (VGKC) antibody is also determined using a similar principle. Briefly, the protein required for the assay is extracted from the cerebral cortex and radiolabelled with a corresponding high affinity ligand, α-dendrotoxin, thus enabling quantification of antibodies against VGKC subtypes KV1.1, 1.2 and 1.6. 23 Similarly muscle (α1) AChR antibodies are often detected using a radioimmunoprecipitation assay involving a radioactive I125 labelled α-bungarotoxin. Muscle specific kinase (MuSK) antibodies are also detected using radioimmunoprecipitation assays.
Review of different antineuronal antibodies
For the purpose of this review, the antineuronal antibodies have been classified based on their antigenic target location – i.e. intracellular or extracellular. They can also be subclassified based on whether the predominant underlying clinical syndrome is paraneoplastic or not (Figure 1).
PNAs against intracellular antigens
There are six such specificities in this group that are widely recognized and have been well-characterized in terms of the target antigens (also referred to as ‘onconeuronal antigens’), with strong links to both cancer and the neurological disorders. Table 4 briefly summarizes the staining patterns and the correlation of six PNAs (in order of their detection frequencies: ANNA-1, CRMP5, PCA-1, amphiphysin, ANNA-2 and Ma2), with the neurological ailments and commonly occurring neoplasms. Most of the PNAs are IgG and are rarely encountered in healthy subjects, 24 but tend to be found in high titres in both the cerebrospinal fluid (CSF) and the serum of patients with PNS. These titres do not correlate with the disease severity but occur at a higher frequency in patients with malignancy and neurological disorder compared with patients with isolated similar cancer. Conversely, lower titres of the antibody can be identified in patients without neurological syndrome; therefore, the presence of the PNA in isolation cannot be used as the only condition for defining the illness as paraneoplastic. 9 However, the diagnostic sensitivity improves enormously when PNAs are used in conjunction with a high index of clinical suspicion.
Examples of staining patterns seen on the cerebellum and the Purkinje cells with (a) ANNA-1, (b) CRMP5, (c) PCA-1, (d) amphiphysin, (e) Ma2 and (f) ANNA-2, and their clinical and predominant cancer associations
SCLC, small cell lung carcinoma; ANNA-1, antineuronal nuclear antibody type 1; ANNA-2, antineuronal nuclear antibody type 2; PCA-1, Purkinje cell cytoplasm antibody type 1; CRMP5, collapsin response mediator protein type 5; Ma2, paraneoplastic neuronal antigen Ma2
Despite the fact that PNAs are sensitive markers of PNS, they may not be detectable in up to 50% of PNS patients (Figure 2). 9 Furthermore, antibodies can be found in up to 16% of neurologically asymptomatic patients with cancer, 24,25 and this is further complicated by the fact that in about 30% of patients with PNS more than one PNA can be identified concurrently. 19 It is also worth noting that in patients with a definite diagnosis of PNS (based on clinical and immunological characterization), the underlying cancer may not be detected for up to 20 months.
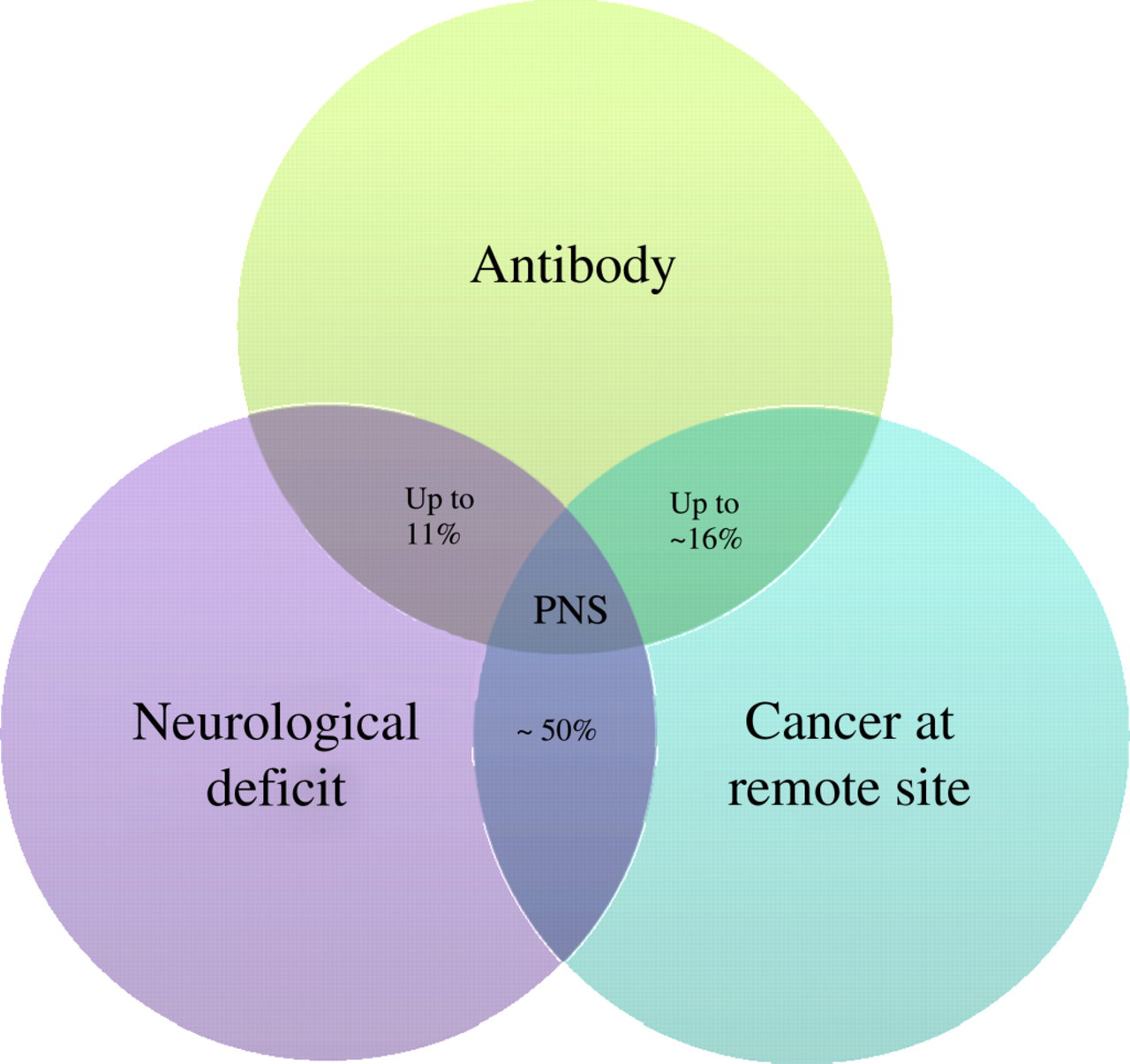
Relationship between neurological deficit, autoimmunity and cancer is complicated by the fact that not all patients with paraneoplastic neurological disorder (PNS) harbour paraneoplastic neurological antibody (PNA). Some patients with cancer and low titre antibody do not present with neurological deficit, while in others cancer cannot be found despite having both PNAs and neurological deficit
Below is an outline of the common PNAs with intracellular targets.
Antineuronal nuclear antibody type 1 (also known as Hu)
ANNA-1 is the most commonly occurring, easily detected and widely investigated PNA that has its antigen located predominantly in both the peripheral and central neurons, and also in tumour cells. The antigen is not seen in glial cells, endothelial cells or non-neuronal tissues (except in the relevant tumour). On a Western blot, the antibody recognizes a protein of molecular size of 35–40 kDa which can be visualized immunocytochemically on primate cerebellum where it binds to the nucleus of the Purkinje cell and reacts weakly with the cytoplasm (Table 4a).
When this antibody is encountered in adult patients who are habitual smokers, it has a very high sensitivity for PNS (>80%) with a common clinical presentation of peripheral neuropathy and underlying small cell lung carcinoma (SCLC). 26 Patients with ANNA-1 are predominantly men in their sixth decade of life with very poor prognosis and, in the majority of cases, have a 60% mortality or severe irreversible disability from their neurological impairments. This antibody can be present at low levels in cancer patients who do not have neurological symptoms. Treatment of the cancer does not always improve neurological function but stabilizes them with limited progression.
Antineuronal nuclear antibody type 2 (or Ri)
The antigenic distribution of ANNA-2 is similar to that of ANNA-1, but confined to the central nervous tissue (Table 4f). Cryosections of stomach containing myenteric plexus were frequently used to distinguish between ANNA-1 and ANNA-2 specificities, particularly prior to the availability of commercial recombinant blots with neuronal antigens. On a Western blot, this antibody recognizes two proteins with a molecular weight of 55 and 80 kDa. Similar to ANNA-1, this antibody can also coexist with other PNAs such as amphiphysin, CRMP5 and ANNA-1. 19
Clinically, the majority of patients harbouring this antibody are in their sixth decade, female, habitual smokers and present with ataxia and ocular movement disorders. 19 The detection of this antibody identifies the neurological problem as being paraneoplastic of autoimmune origin and the antibody is predominantly associated with breast cancer and SCLC.
Antineuronal nuclear antibody type 3
The rarer antineuronal nuclear antibody type 3 (ANNA-3) has been described in a few patients with PNS associated with SCLC, and stains the nuclei of the Purkinje cells strongly, with minimal staining of the molecular layer and without staining of other cerebellar neurons. 27 The antibody can stain the nuclei of renal podocytes and has a molecular weight of 170 kDa on Western blotting. Like the other ANNAs, they can coexist with other PNAs.
CRMP-5 or CV2 antibody
The antibody is known by two names, CRMP5 and CV2, which are used interchangeably. The specific antigen recognized by the CRMP5 antibody is the cytosolic phosphoprotein with a molecular size of 66 kDa, known as collapsin response mediator protein type 5 (hence the name, CRMP5), that is located in a subpopulation of oligodendrocytes, a subset of sensory peripheral neurons, Schwann cells and SCLC cells. Its antigen is relatively difficult to detect as it can be confused with non-specific astrocyte staining that is often seen with most samples (Table 4b).
Anti-CRMP5 antibody is the second most frequently occurring PNA that shares some common features with ANNA-1 in terms of the clinical picture and neoplasm. Like ANNA-1, it is also linked to a diverse range of neurological syndromes, with peripheral neuropathy being the most frequent, with an underlying SCLC occurring at a higher frequency of about 80%. 28 However, there are also some significant differences between the two antibodies, especially in terms of median survival time which is considerably longer (48 months) in patients with CRMP5 antibody compared with patients with ANNA-1 antibody (11 months). In patients with coexistent CRMP5 and ANNA-1 antibodies, the median survival is around 18 months. 29
Purkinje cell antibody, PCA-1 (Yo)
PCA-1 is the third most commonly detected antibody whose target proteins (molecular size of 34 and 62 kDa) are highly concentrated in the cerebellar Purkinje cell cytoplasm. The distribution can be seen as a characteristic coarse staining of the Purkinje cell cytoplasm (Table 4c) which is due to its binding to ribosomes, endoplasmic reticulum and Golgi complex vesicles. The presence of PCA-1 antibody displays tight homogeneity and very strongly correlates with patient gender and type of cancer. Oligoclonal bands have also been demonstrated in the CSF of patients with PCA-1 antibody (Figure 3). This antibody occurs almost exclusively in female patients with paraneoplastic cerebellar degeneration and breast or ovarian cancer; conversely, only a few isolated cases of this antibody found in men have been documented. Unlike ANNA-1 or ANNA-2, PCA-1 is almost always exclusively detected alone (without any other PNAs). One-third of patients develop severe progressive neurological impairment leading to death, while in around half of the patients, the death is due to the tumour itself. 30 Unfortunately, treatment has little or no effect on the neurological syndrome. It is important not to confuse PCA-1 with other rare paraneoplastic antibodies that are cytoplasmic such as Tr or Purkinje cell antibody type 2 (PCA-2), as their clinical and tumour associations are different. 31

Oligoclonal bands seen in the cerebrospinal fluid (CSF) only. This demonstrates intrathecal IgG synthesis in a 48-year-old woman with cerebellar ataxia harbouring Yo antibody
Purkinje cell antibody type 2
PCA-2 antibody has been identified in a small group of patients in the USA but confined to SCLC and is identified by their binding to the cerebellar Purkinje cell soma and dendrites, internal granular layer and dentate nucleus. 31 They also stain the neural elements in the kidney and gut and Western blots reveal a 280-kDa band.
Anti-Tr antibody
The Tr antibody (also called PCA-Tr antibody) is linked predominantly to men with paraneoplastic cerebellar degeneration and Hodgkin's disease. 32 Tr antibody has been partially characterized but its identification remains dependent on the immunocytochemical fine speckled distribution pattern in the cytoplasm and proximal dendrites of Purkinje cells. Attempts to detect it by a secondary system such as Western blot have not been very successful.
Antiamphiphysin antibody
Antiamphiphysin antibodies recognize a 128-kDa protein that is widely distributed throughout the central and peripheral nervous system. The antigen is expressed in the neuropil, a region where synaptic connections are formed between branches of axons and dendrites (Table 4d). The granular layer of the cerebellum is rich in these interneuronal connections, containing a high density of neuropil where amphiphysin can be located immunocytochemically. The binding of amphiphysin to cerebellar structures produces a characteristic staining pattern that has a similar distribution pattern as glutamic acid decarboxylase (GAD) antibody which can be resolved by testing the sample on pancreas that is rich in GAD enzyme or using recombinant proteins.
In 74% of patients, amphiphysin has been detected in conjunction with other PNAs such as ANNA-1, ANNA-2, PCA-2 or CRMP5. 33 Like many of the PNAs, the immunological basis for aetiology of the neurological disorder is evident from abnormalities seen in the CSF and the brain: postmortem studies have shown the presence of inflammatory mediators (CD8+ T-cells). It must also be emphasized that like ANNA-1 and CRMP5, amphiphysin can also be present in neurologically asymptomatic patients with cancer. 24
Amphiphysin occurs at a very low frequency of 0.06% in patients with a mean age of 64 years (M:F ratio 1:1.52) and as mentioned above, present with a wide spectrum of neurological manifestations, with neuropathy being the most common, followed by encephalopathy, myelopathy, cerebellar and SPS. The multifocal nature of the neurological disorder is likely to be due to targeting of multiple antigens which is seen in the majority of patients. In 86% of men, SCLC was the most common neoplasm. However, a third of patients with isolated amphiphysin specificity were women with breast cancer, suffering from SPS or myelopathies. 33 Treatment has no statistically significant effect on the survival time for patients compared with those who did not receive any specific therapy.
Anti-Ma2 (also referred to as Ta) antibody
Anti-Ma antibodies react with the Ma1, Ma2 and Ma3 proteins and also cross-react with a variety of tumours. Of the three, paraneoplastic association with anti-Ma2 antibodies is the most common. The target antigen is located in the neuronal nucleoli of the central nervous system, and to a lesser extent, in the nucleus and cytoplasm. Ma1 protein is found in both testicular germ cells and central neurons while Ma2 is specific to the central nervous system and Ma3 has also been found in several systemic tissues. In terms of detection, non-Ma nucleolar staining (either cross-reactivity from antinuclear antibodies or species-specific) can be often encountered on the primate tissue which can be resolved with further testing using specific Ma2 antigen. 20 Ma antibody can be visualized as a single dot in the nuclei of cerebellar Purkinje cells (Table 4e) with some weak staining in the cytoplasm.
Patients with Ma2 antibodies tend to be young male adults (mean age 46 y) with the majority suffering from limbic encephalitis (LE) and germ-cell tumours of the testis. 34 Improvement or stabilization of the neurological syndrome is seen in two-thirds of patients with anti-Ma2 antibodies but only in about a fifth with other members of the Ma family such as Ma1. 34 Death will occur in fewer than 15% of all these patients within a year of diagnosis, but, in most cases, from neurological deterioration.
Ma1 and Ma3 are confined to older patients presenting with more diverse and severe symptoms of the cerebellum, brainstem and limbic system. The majority of the patients suffer from large cell lung tumours, and less commonly, gastrointestinal and breast malignancies. 34
Antiglial nuclear antibody (SOX1 antibody)
Antiglial nuclear antibody (AGNA) has been recently described and differs from the other paraneoplastic antibodies mentioned above in that they are non-specific (i.e. they can occur with the neurological syndrome or the tumour in isolation) and intrathecal IgG synthesis cannot be demonstrated. 35 SOX1 (sex determining region Y – Box 1) has been identified as the corresponding antigen for AGNA that is found in the nuclei of Bergmann glial cells of adult cerebellum (Figure 4). SOX1 belongs to a family of DNA-binding transcriptional factors and the gene encoding SOX is classified into eight subgroups labelled A to H. SOX1, SOX2 and SOX3 proteins belong to group B1 and are expressed in both the adult and developing nervous systems and also in SCLC.
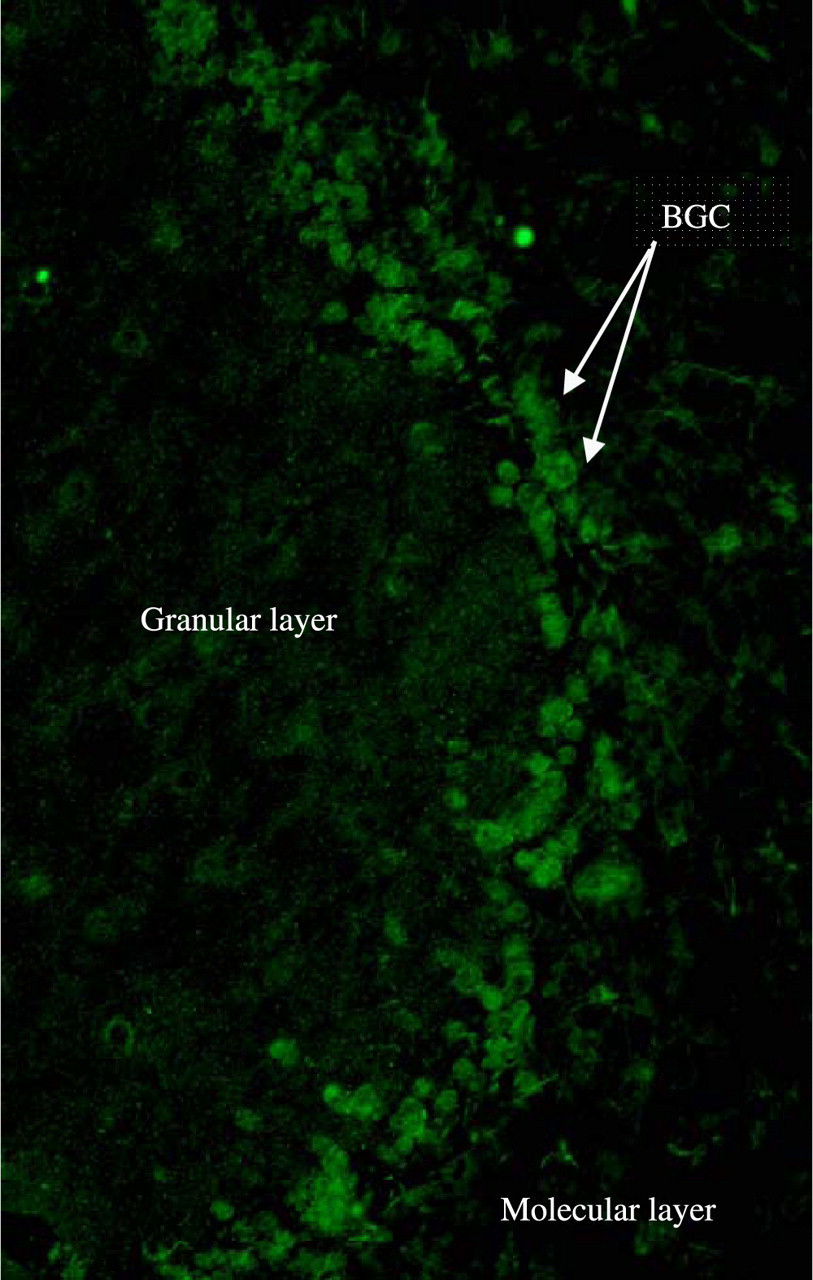
Antiglial nuclear antibody (AGNA) antibody from a 66-year-old man, staining the nuclei of Bergmann glial cells (BGC) on the border of the molecular layer and the Purkinje cell layer of primate cerebellum
SOX1 in isolation does not appear to be cancer-specific but when it coexists with VGCC antibody causing LEMS, it then predicts the presence of SCLC. This is evident from a recent study which found SOX1 antibodies more frequently in patients with paraneoplastic LEMS (64%) caused by VGCC compared with idiopathic LEMS or SCLC alone (22%). 35 The usefulness of SOX1 antibody is apparent in distinguishing between idiopathic (no tumour found) and paraneoplastic LEMS (where SCLC is present), since these neurological conditions cannot be clinically differentiated. The presence of this antibody should alert the clinician to search for an underlying neoplasm (SCLC), in patients with LEMS 35 and in cases where a tumour has not been found, regular monitoring of the patient for a neoplasm is advised.
Anti-Zic4 antibody
Anti-Zic4 antibody targets Zic4 protein belonging to a family of five zinc-finger proteins (Zic1, Zic2, Zic3, Zic4 and Zic5) encoded by the Zic gene family. ZIC4 gene is expressed in adult cerebellum and works cooperatively with the other Zic genes during cerebellar development, 36 and any experimental mutation of the Zic gene compromises normal neurological function.
Like AGNA, this occurs less commonly and is non-specific with regard to their immunopathogenic role. An isolated Zic4 antibody response in patients does not have a significant association with either the cancer or the neurological deficit, as the incidence of pure Zic4 immune response in patients with neurological disorder (predominantly cerebellar) and SCLC was less frequent (15%) but similar to the incidence of Zic4 antibody in SCLC alone. 37
Zic4 antibody, in isolation, reacts predominantly with the nuclei of cerebellar granular cells. It is very rare to encounter isolated Zic4 in the laboratory, as in the majority of the cases (over 80%), Zic4 coexists with other SCLC-associated PNAs (such as ANNA-1 and CRMP5). Immunocytochemical visualization will be dominated by the co-expressing onconeuronal antigen (an expression coined for paraneoplastic antigens) and overshadowed by the commonly occurring PNAs when tested on line blot as these do not contain antigens for Zic4.
The coexisting antibodies reflect the diversity of the immune response which is mirrored by a wide range of neurological symptoms. In cases where Zic4 is specifically identified, it predicts the presence of a neoplasm, usually SCLC (92%), and suggests that the neurological ailment is likely to be paraneoplastic.
Non-paraneoplastic antibodies against intracellular antigens
GAD antibody
GAD antibody is associated with type 1 diabetes mellitus (GAD65) and neurological deficiencies (GAD67) that are mainly of non-paraneoplastic origin. These two isomers share a high degree of sequence homology and have a molecular size of 65 and 67 kDa, respectively. Currently, most assays available in the UK are unable to distinguish one isoform from the other and a detailed discussion on the immunology of type 1 diabetes mellitus is beyond the remit of this review. GAD antibodies share some common characteristics with amphiphysin in that they both have a similar immunocytochemical distribution in the cerebellar granular layer and can be clinically associated with SPS. In our laboratory, GAD antibodies are routinely measured by a commercial ELISA kit and less frequently using the immunocytochemical method. In neurological disorders, GAD antibodies are expressed as an index which is calculated using the concentration ratio of GAD in paired CSF and serum samples divided by albumin CSF:serum ratio. 38 Among the patients with an index greater than 1, inferring intrathecal GAD synthesis, 100% presented with cerebellar ataxia, 85% had SPS and 86% suffered from other neurological disorders. In contrast to amphiphysin, GAD antibodies, in most cases, are not associated with underlying malignancy and are considered as non-paraneoplastic antibodies. However, there are few exceptions where GAD is recognized as a paraneoplastic phenomenon occasionally in patients with LE, paraneoplastic encephalomyelitis and paraneoplastic cerebellar degeneration. 38
Adenylate kinase 5 antibody
Antibodies to adenylate kinase 5 have been reported to be associated with LE without any underlying neoplasm. 39 These patients are poorly responsive to immunomodulatory treatment.
Homer 3 antibody
Homer 3 is expressed in the Purkinje cells and specifically binds to the C termini of the metabotropic glutamate receptors, mGluR1. An isolated case report of Homer 3 antibody in subacute idiopathic cerebellar ataxia has been published in the literature. 40
Antibodies to extracellular antigens
This section deals with the aspects of pathogenic antibodies that exert their effects by binding to extracellular antigens, thus producing disorders of the nervous system. Several antibodies fall in this category, notably: VGKC, NMDA, alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), gamma aminobutyric acid type B (GABAB) and glycine receptors. Characteristically, their mode of action, association with neurological condition, method of detection and response to treatment, appear to be similar. Intrathecal synthesis has been demonstrated in most of these antibodies and their titres have been shown to correlate with the disease state, suggesting immune-mediated pathology. Most of these antibodies were initially described to be purely of paraneoplastic origin; however with the increased detection of these, most patients do not seem to have an underlying neoplasm. Essentially, these antibodies are markers of neuronal surface antigens.
The majority of these antibodies have been linked to LE, an inflammatory neurological condition, predominantly affecting the hippocampus and amygdala, resulting in neuropsychiatric disorders. Hallucinations, REM sleep and autonomic dysfunction and peripheral nerve hyperexcitability are frequently encountered in affected patients. The changes in the behaviour and mood are noticed by the irritability, depression, sleep disturbance, seizures and short-term memory loss.
The cell surface antigens are polypeptides with their antigenic epitopes exposed on the external surface of the pre- or postsynaptic terminals and functionally these are engaged in synaptic transmission. They are also known as receptors, composing of several subunits which house binding sites for natural compounds such as glutamate (a neurotransmitter), dietary toxins and therapeutic agents. A number of processes including autoimmune response against receptor subunits can interfere with signal transduction and influence synaptic transmission.
VGKC antibody complex
VGKC is a member of the Shaker family of potassium channels found both in the central and peripheral nervous systems. This transmembrane protein comprises of three important α-subunits (KV1.1, KV1.2 and KV1.6) that form the channels specific for potassium movement and plays a crucial role in depolarizing the cell to a resting state. 41 It also has a high affinity for green mamba snake venom (α-dendrotoxin) and it is this property that is utilized for determining anti-VGKC antibodies, in the radioimmunoprecipitation assays.
Contrary to the previously held belief, data from recent studies imply that these autoantibodies may be directed against complexing proteins associated with VGKCs, such as contactin-associated protein 2 (Caspr-2), leucine-rich glioma inactivated protein 1 (Lgi-1) and Tag-1/Contactin-2. 42,43 High titres of VGKC antibodies can be found in the serum of patients with LE and the cellular expression may be visualized on the cerebellum as having a characteristic staining in the molecular layer with the Purkinje cell cytoplasm and all other cerebellar nuclei being spared. 23 However, routinely, VGKC antibodies are measured using a radioimmunoassay.
Antibodies to VGKC have been described predominantly in men (90%) with non-paraneoplastic classical LE and Morvan's syndrome, 23 but data emerging in a later publication using a cohort of 80 patients linked VGKC antibodies to a broader spectrum of neurological manifestations and malignancies than previously appreciated. 44 The most frequent neurological manifestations can be traced to the involvement of various areas of the CNS, including the cerebral cortex (affecting cognitive impairment [71%], seizures [58%] and hallucinations [10%]), hypothalamus (38%), extrapyramidal system (21%), autonomic pathway (33%) and the peripheral nerves (25%). In the later situation, 17% of patients suffer from hyperexcitability of motor nerves (neuromyotonia), leading to muscle twitching, cramps and weakness, caused by an increased release of acetylcholine into the synaptic cleft by VGKC antibody-mediated prolonged action potential. 41 Recent studies have shown that patients with antibodies directed against the Lgi-1 protein predominantly have an LE phenotype whereas the Caspr-2 antibodies were more likely to produce neuromyotonia or Morvan's syndrome. It is also thought that the presence of Caspr-2 antibodies was more likely to indicate an underlying neoplasia and poorer prognosis. 42 Recent studies have also shown that Caspr 2 antibodies may also be seen in patients with subacute cerebellar ataxia using a cell-based assay, and may not always be detectable by standard VGKC radioimmunoprecipitation techniques. 45
Additional symptoms were also observed in patients where VGKC coexisted with other antibodies, for example, diabetes mellitus due to GAD autoimmunity and myasthenia gravis because of AChR antibody. 44 Tan et al. found that 47% of the patients with VGKC antibodies (majority being smokers) also had a range of cancers which included SCLC (7%), thymoma (6%), prostate adenocarcinoma (6%) and adenoma (7%).
VGCC antibody
VGCC antibody binds to extracellular antigens and is directed against the P/Q type of calcium channels, expressed in the cerebellar molecular, Purkinje and granular cell layer but predominantly found in the cytoplasm of Purkinje cells and SCLC. 46 P/Q channels are sensitive to ω-conotoxin, a neurotoxin from the marine cone snail and this property of the VGCC channels is utilized for quantifying anti-VGCC antibodies. Radioiodine neurotoxin (125I-conotoxin) is used for labelling the voltage-gated P/Q-channels. The precipitated radiolabelled-receptor–antibody complex is quantified. 22
Autoimmunity to the presynaptic P/Q type of VGCC is responsible for the decrease in acetylcholine release leading to skeletal muscle weakness and autonomic symptoms seen in the neuromuscular disease known as LEMS. Approximately half of the patients have a paraneoplastic form of LEMS, the majority being men, habitual smokers with usually SCLC as an underlying neoplasm. 47 Similarly, a rapid onset of the disease, with involvement of the upper limbs and distal muscles, has been shown to be associated with an underlying SCLC. 48 These patients tend to respond well when the cancer is treated, leading to better prognosis than LEMS without cancer. If no tumour is found, regular monitoring for occult malignancy is recommended, particularly in older patients (>50 y) with a history of long-term smoking and/or evidence of coexisting autoimmune disease. As mentioned previously, the presence of AGNA (SOX1) antibodies in patients with LEMS is more closely linked to an underlying SCLC.
Glutamate receptor antibodies
Glutamic acid is a member of the excitatory amino acid neurotransmitters, referred to as glutamate, found in the CNS and is involved in numerous brain functions such as cognition, memory, learning, movement and development. As a neurotransmitter, it functions by binding to postsynaptic glutamate receptors (GluR), thereby facilitating the propagation of the nerve impulse across the synapse between the two nerve terminals. 49 About 50% of all the synaptic transmission in the CNS is thought to be mediated by glutamate via two types of glutamate receptors (ionotropic [iGluR] and metabotropic [mGluR]) found on both the pre- and postsynaptic membranes. There are two types of iGluRs, acquiring their names from high-affinity compounds, NMDA and AMPA. These receptors are ionic channels allowing sodium ion influx and membrane depolarization on stimulation with glutamate (Table 5). Deviation from normal receptor activity, particularly hyperactivity, is considered to cause neurotoxicity (also referred to as excitotoxicity) which can result in severe neuronal damage and maybe responsible for pathogenesis of acute and chronic insults to the CNS. 49
Types of macromolecules involved in the excitatory and inhibitory synaptic transmission
GAD, glutamic acid decarboxylase; Na+, sodium ion; GABA, γ-aminobutyric acid; Cl−, chloride ions; GluR, glutamate receptor; Ca2+, calcium ions; NMDA, N-methyl-
NR are subunits of the NMDA receptor
*AMPA and Kainate are similar to NR subunits
Both NMDAR and AMPA receptor (AMPAR) antibody share common features and are considered pathogenic. Both can be associated with malignancy and their CSF antibody titres correlate with the neurological outcome, response to immunotherapy and treatment of the underlying tumour. These observations suggest a pathological role of the antibodies in the aetiology of the disease. From the clinical point of view, the presence of these antibodies indicates a possibility of underlying neoplasm, most commonly lung, breast or thymus. However, these antibodies can also occur without neoplastic condition.
NMDAR antibody
NMDAR comprises four NR units, each subunit has a molecular size of about 100 kDa and together they form an ion channel/receptor. The two NR1 units make up a binding site for glycine while the other two NR2 (A, B, C or D) subunits bind glutamate. Activation of the NMDAR facilitates an increase in intracellular Ca2+ ion initiating a cascade of cellular events. 49 It has been suggested that NMDAR antibodies inhibit the receptors on the presynaptic GABAergic interneurons, resulting in reduction of GABA release, thereby diminishing inhibition of the postsynaptic glutamatinergic transmission. The consequent excessive release of glutamate in various structures of the brain causes neuronal excitotoxicity. 50
Cellular distribution of the NMDAR antibodies can be ascertained on non-permeabilized hippocampal neurons or the cerebellum where the NMDAR antibody binds to the neuropils in both the molecular layer of the hippocampus and the granular layer of the cerebellum (Figure 5a). The NMDAR antibodies can also be determined with much higher sensitivity and specificity using NR1-transfected cells (Figure 5b). These cells are now commercially available for detecting NMDAR antibodies. As yet, there are no commercial Western blots to complement immunocytochemical detection.
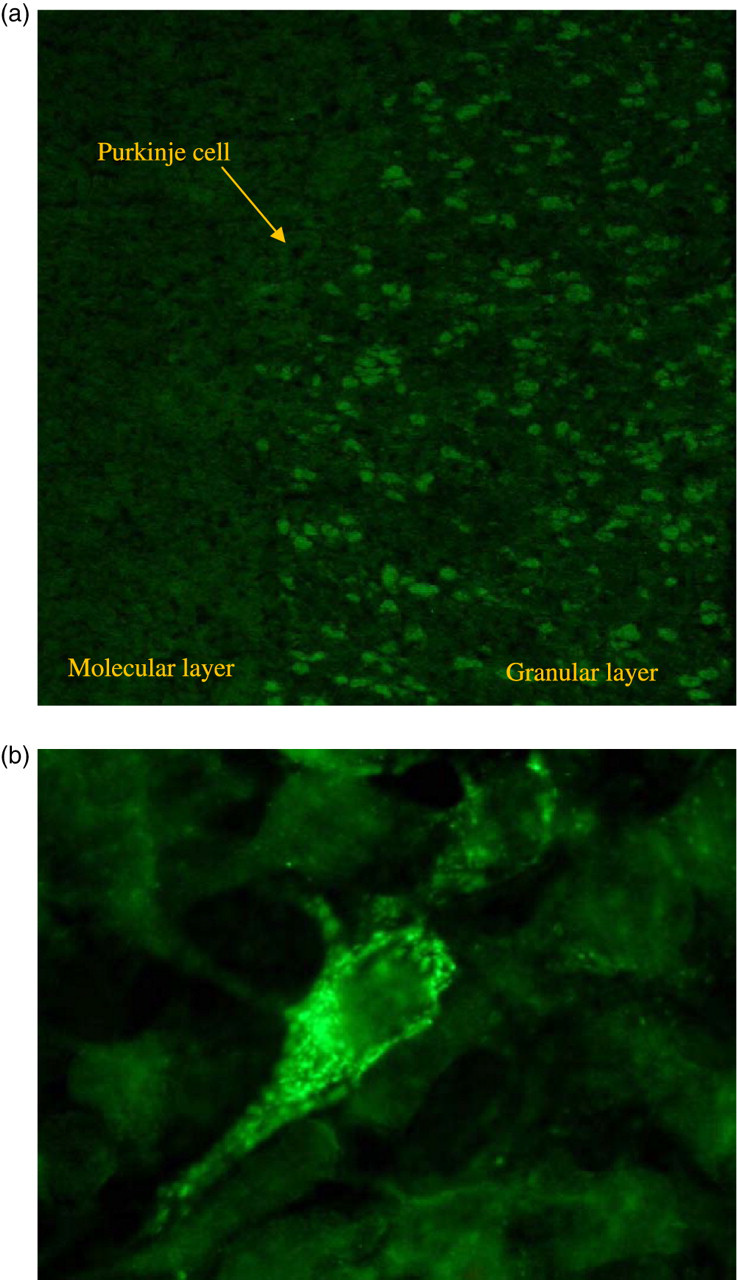
(a) Staining of primate cerebellar granular layer with N-methyl-
Recently, autoimmunity to the NR1 subunit of the NMDAR has been identified in 100 patients with severe but treatment-responsive LE with prominent psychiatric presentation, involuntary movements, autonomic symptoms and respiratory depression. 21 This syndrome was initially described in young women over the age of 18 y with ovarian teratoma (56%) but has also been reported at a lower frequency (9%) in girls under the age of 14 y. Most recent observations suggest that the spectrum of the disease is expanding to include non-neoplastic cases and it is no longer confined to the female gender. Furthermore, varying levels of NMDAR antibodies are found in different subset of patients. For instance, the levels of NMDAR antibody in the non-paraneoplastic patients are significantly lower than that found in the paraneoplastic syndrome. 51 There appear to be two phases of the disease, the early one is linked to the CSF lymphocytosis and the later phase seen with the appearance of CSF oligoclonal bands, but in any case the treatment appears to be more effective in the early stages whether it is immunotherapy or tumour removal. 51
AMPAR antibody
Like NMDAR, AMPAR is an ionotropic transmembrane receptor for glutamate that is positioned on the postsynaptic membrane and is composed of four subunits (GluR1, GluR2, GluR3 and GluR4) that host four binding sites located on each unit. 49 In response to stimulation of the AMPAR by the natural neurotransmitter glutamate, sodium influx leads to depolarization of the postsynaptic cell, thus contributing to the fast synaptic transmission observed in the CNS. The GluR1 and GluR2 subunits are responsible for these events.
However, in the presence of the AMPAR antibody, the number of the GluR2-AMPAR clusters at the synapse is reduced, which is caused by the disruption of the receptor trafficking/turnover leading to the clinical manifestation. 52 Antibodies to GluR1/2 subunits of the AMPAR have been described in 10 female patients with autoimmune LE. The majority of the patients had an underlying malignancy of the thymus, breast or lung. 52 The noticeable clinical feature of both NMDAR antibody and GluR1/2 autoimmunity was a tendency for the patients to relapse.
Similar to the NMDAR assay, HEK293 cells expressing GluR1/GluR2 receptors have been used for diagnostic testing of these antibodies in serum samples or CSF.
mGluR1 antibody
The mGluRs are a family of eight single polypeptide chain receptors that bestow their function through a second messenger (G-protein), modulating the channel and enzyme function. 49 Only a handful of patients have been described to harbour this antibody, manifesting as cerebellar ataxia in the presence of Hodgkin's lymphoma.
Antibodies to GluR3 were thought to be associated with Rasmussen's encephalitis, which is characterized by progressive focal epilepsy, hemiparesis and unilateral brain atrophy. 53 However, further studies have shown that these antibodies are relatively non-specific and may not be seen commonly in patients with this condition, thereby having doubtful clinical significance. 54–56
GABAB receptor antibody
Antibodies against the B1 subunit of the inhibitory GABAB receptor have been associated with seizures and cognitive changes in patients with LE, half of which were associated with an underlying tumour, commonly SCLC. 57 This cell-based assay is performed using transfected B1 and B2 subunits of the GABAB receptor on HEK-293 cells. In a larger series of 65 LE patients, seven (11%) had GABAB antibodies, with five of them associated with SCLC. 58
Glycine receptor antibody
The inhibitory glycine receptors (GlyR) are mainly concentrated in the brainstem and spinal cord. Mutations of the α1 subunit of GlyR have been known to be associated with hereditary hyperekplexia, an exaggerated startle syndrome. Recent observations have shown that antibodies against this subunit can be associated with autoimmune hyperekplexia, SPS and progressive encephalomyelitis with rigidity and myoclonus. 59 These antibodies have also been identified using a cell-based assay in a large group of patients with similar symptoms, with very high levels of intrathecal synthesis. 60
Other antibodies in neurological diseases
A detailed description of all other antibodies associated with neuroimmunological diseases is beyond the scope of this review. The more common ones are mentioned in Figure 1 and Table 1. Of these, aquaporin-4 antibodies seen in neuromyelitis optica can be detected by several techniques with varying sensitivities (in brackets): indirect immunofluorescence (86%), cell-based assay (91%) and fluorescence-based immunoprecipitation assay (83%). 61 The cell-based assays use either or both of the M1 or M23 isoforms of aquaporin-4, with those using the M23 isoforms having a slightly higher sensitivity. 62 Similarly, muscle (α1) AChR antibodies are often detected using a radioimmunoprecipitation assay involving a radioactive I125-labelled α-bungarotoxin (from the banded krait), but can also be detected by a sensitive, non-isotopic ELISA technique, 63 and are seen in approximately 85% of patients with generalized myasthenia gravis, characterized by muscle fatigability. Some low-affinity AChR antibodies can only be detected using a cell-based immunofluorescence assay where the AChRs are clustered on HEK cell surfaces using Rapsyn. 64 A proportion of patients without AChR antibodies have antibodies against a different muscle membrane protein called MuSK. 65 MuSK antibodies are also detected using a radioimmunoprecipitation technique. Acetylcholine receptors found in the autonomic ganglia are prone to immune attack in approximately 50% of patients with autoimmune autonomic gangliopathy (AAG), causing postural hypotension, impotence, bowel/bladder disturbances and impairment of sweating, salivation or lacrimation. 66 Fifteen percent of patients with AAG have an underlying tumour, commonly SCLC or thymoma. 67 The radioimmunoassay for detecting α3 neuronal AChR antibody is similar to that for the more common antibodies against the muscle AChRs, but using I125-labelled epibatidine (from the frog, Epipedobates tricolor) and solubilized human neuroblastoma AChRs. Antiganglioside antibodies have been detected in a variety of inflammatory neuropathies with different clinical phenotypes and can be directed against several individual gangliosides (GM1, GD1a, GQ1b, etc.) or against ganglioside complexes (e.g. GM1 complexed with GD1a). 68,69 They are detected using ELISA and multiplex glycoarrays. 70 Antibodies against myelin-associated glycoprotein have been found in approximately 50% of patients with an IgM paraproteinemic demyelinating neuropathy. 71 The antibodies which are usually IgMκ are detected by ELISA or by immunofluorescence on peripheral nerve sections (Figure 6). Antibasal ganglia antibodies (BGA) are cross-reacting antibodies, triggered by a Group B streptococcal infection, which have been implicated in a group of conditions encompassed under the term, paediatric autoimmune neuropsychiatric disorders associated with streptococcal infection (PANDAS), which could manifest as chorea (a rhythmic movement disorder), obsessive compulsive behaviour or tics (causing sudden repetitive non-stereotyped movements or vocalization). BGA have been described in this group of neurological conditions by ELISA and Western blotting, 72 but this has recently been challenged in other studies. 73,74
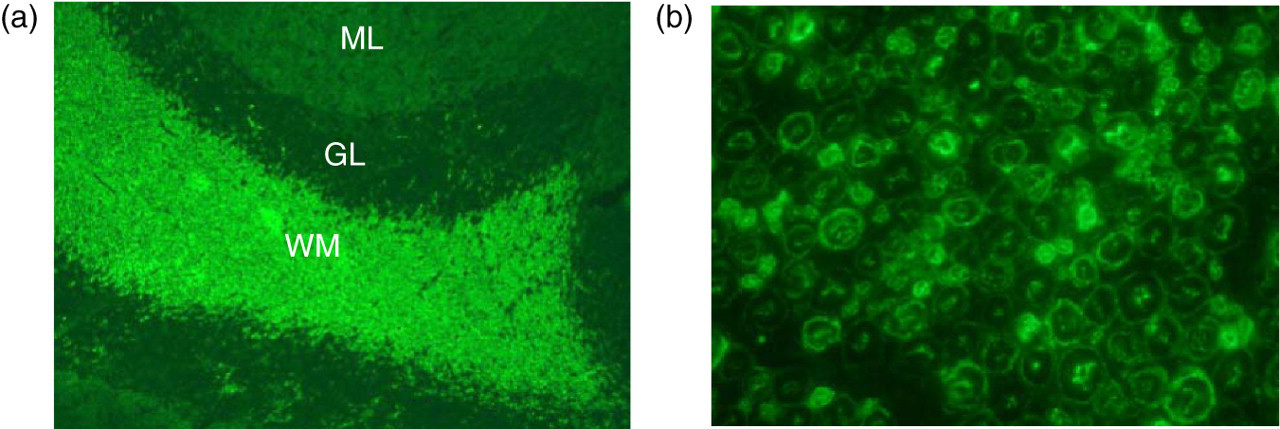
IgM anti-myelin-associated glycoprotein (MAG) antibodies reacted with primate cerebellum (a) and the sciatic nerve (b). The MAG antibody predominantly recognizes the myelin bundle in the white matter (WM) of the cerebellum, leaving the granular (G) and the molecular (M) layers (L) unstained, whereas in the sciatic nerve, MAG staining lights up the inner and outer myelin layers of the axons
Conclusions
The multifaceted nature of the syndromes associated with these antibodies is often related to the diversity of the immune response against neuronal tissue. Even in the absence of a clearly defined antibody, clues pointing to an autoimmune aetiology may be obtained by observing CSF pleocytosis, elevated protein and in about 30%, by the presence of intrathecal IgG synthesis in the form of oligoclonal bands.
For PNS, good practice guidelines from the European Federation of Neurological Societies (EFNS) Task Force, recommend a focused tumour search based on the underlying onconeuronal antibody. 75 Currently, early treatment of the tumour seems to offer the best approach for stabilizing PNS. 76 Patients who are treated for cancer should be regularly monitored for complete blood count, liver enzymes and renal function in order to determine toxicity to therapy. This could also include serum paraneoplastic antibody titres in selected syndromes. 77 In many cases, even after the initial search for malignancy has been exhausted and a tumour has not been found, the patient is monitored every six months for up to four years in order to search for the cancer. 75
Most of the recently described syndromes associated with neuronal cell surface antibodies are treated effectively by immunomodulatory therapy with intravenous immunoglobulins or plasma exchange. Some of these antibodies may also be associated with tumours and the timely identification and removal of these offers the best long-term prognosis.
Antibodies directed against the neuronal proteins are of importance in defining the type of disease whether PNS or non-PNS, its responsiveness to immunotherapy and disclosure of a previously unknown underlying malignancy. Therefore, early recognition and accurate diagnosis followed by an appropriate therapy can improve neurological function, enhance the quality of life and possibly contribute to survival.
DECLARATIONS