
Abstract
We present the case of a 57-year-old patient who initially presented with a constellation of symptoms including intense pruritis, flushing and diarrhoea. Following several months clinical deterioration, the patient was investigated radiologically, where multiple hepatic tumours were identified. Liver biopsy confirmed the presence of a well-differentiated metastatic gastroenteropancreatic endocrine carcinoma with biochemical evidence of serotonin secretion. Over a period of six months, the clinical course of the patient's disease progressed whereby severe hypoglycaemia became the major manifestation. Subsequent biochemical investigations confirmed the diagnosis of an insulinoma. Extensive radiological investigation revealed a solitary primary pancreatic tumour, indicating the presence of a metastatic pancreatic endocrine tumour (PET) secreting both insulin and serotonin. The patient was treated with a chemotherapy regimen consisting of 12 cycles of 5-fluorouracil/oxaliplatin, responding clinically – improved World Health Organization performance score from 3 to 1, biochemically – significantly reduced plasma chromogranin A and cancer antigen 19-9 concentrations and improved liver function tests, and radiologically – reduced pancreatic and hepatic tumour size. This is the first report of a primary PET secreting insulin and serotonin. Due to the association of serotonin-secreting gastroenteropancreatic endocrine tumours (GEP-ETs) with multiple endocrine neoplasia type-1 (MEN1) and biochemical evidence of an insulinoma, MEN1 should also be considered in such cases. The case provides further evidence for the biological heterogeneity of GEP-ETs and the myriad secretory humoral products and resultant clinical syndromes arising from such tumours.
Introduction
Gastroenteropancreatic endocrine tumours (GEP-ETs) arise from the diffuse endocrine system of cells. This is the collective term given to endocrine cells located throughout the gastrointestinal mucosa and pancreas. These cells express common markers of neuroendocrine origin, including chromogranin A, synaptophysin and neuron-specific enolase. At least 15 different cell types have been identified, capable of secreting over 40 bioactive peptides, amines and prostaglandins, resulting in a heterogeneous array of clinical syndromes. 1
The carcinoid syndrome is the term applied to the clinical manifestation of serotonin (5-hydroxytryptamine)-secreting ETs. Metabolism of tryptophan, the chemical precursor to serotonin, is altered in the majority of these patients; normally, less than 1% of dietary tryptophan is converted to serotonin; however, this may be up to 70% in patients with the carcinoid syndrome. Serotonin is subsequently metabolized to 5-hydroxyindoleacetic acid (5-HIAA), which is excreted in the urine. Excessive production of serotonin is thought to be the cause of diarrhoea, a common symptom of the syndrome, while secretion of polypeptides, including kallikrein and tachykinins, may account for the characteristic cutaneous flushing. Long-term effects include pulmonary hypertension and cardiac valvular lesions. 2
Fasting hypoglycaemia is the clinical hallmark associated with insulin hypersecretion from pancreatic insulinomas. This may be accompanied by episodes of neuroglycopenic symptoms including confusion and personality change, and autonomic symptoms including palpitations, tachycardia, sweating and nausea. Biochemical diagnosis is based on the demonstration of inappropriately detectable/elevated insulin during hypoglycaemia.
ETs arising from the pancreas may be sporadic or part of the multiple endocrine neoplasia type-1 (MEN1) syndrome. They are capable of producing multiple secretory humoral factors, often being detected late in the course of disease when metastasis has already occurred. 3 Herein, we report a case of a patient with a primary pancreatic endocrine tumour (PET) with extensive liver metastases. The patient originally presented with features of the carcinoid syndrome, with the disease rapidly progressing to that of a severe hyperinsulinaemic hypoglycaemic syndrome. Biochemical evidence of both insulin and serotonin secretion indicated a PET secreting a rare combination of humoral factors.
Case report
We report the case of a 57-year-old patient who presented to their general practitioner (GP) with a 14-month history of intense pruritis following showering, occasionally associated with sweating and skin tingling. The itch was mainly on the shoulders and torso but was not associated with a rash, and there was minimal pruritis at other times. The patient had changed shower gels and soaps, and had self-medicated with over-the-counter antihistamines, but to no avail. An ex-smoker and occasional drinker, there was a past medical history of asthma and no significant family medical history. At the time of initial presentation, the patient was being prescribed budesonide and salbutamol to treat asthma, and omeprazole to treat gastric acid reflux. Routine blood tests including urea and electrolytes, liver function tests (LFTs), full blood count (FBC), lipid profile and overnight fasting glucose were all within reference ranges. The GP prescribed an aqueous cream and referred the patient to a hospital outpatient dermatology clinic.
Eight weeks later in the dermatology clinic the patient described similar symptoms, including worsening pruritis, as well as occasional flushing and tachycardia. The consulting clinician diagnosed aquagenic pruritis, prescribed fexofenadine and dexamethasone and advised the patient to use an aqueous cream in the shower. Blood tests including FBC, erythrocyte sedimentation rate, immunoglobulins and autoantibodies were all within reference ranges.
Eight months following initial presentation, the patient returned to their GP and was investigated by a different physician. The patient had continued to feel unwell, still experiencing episodic flushing and tachycardia and had stopped taking medications due to the development of additional symptoms including diarrhoea, malaise, trembling and muscle fatigue. The patient had also begun to experience symptoms of light headedness, which were relieved by the ingestion of sugary drinks. On examination, the liver was palpable and a blood test revealed abnormal liver function – alkaline phosphatase (ALP) 254 U/L (30–130), alanine aminotransferase (ALT) 139 U/L (1–41) and overnight fasting glucose 2 mmol/L (3.5–6.0). The GP made a referral for an abdominal ultrasound scan.
Two weeks later the patient underwent radiological investigation where, on examination, the liver remained enlarged. An ultrasound scan suggested the presence of multiple hepatic tumours, which was subsequently confirmed by a computerized tomography (CT) scan. Following hospital admission, a tumour marker panel was requested – cancer antigen 19-9 (CA19–9) 280 U/L (<35), alpha fetoprotein 9 U/L (<10) and carcinoembryonic antigen <3 μg/L (<7). On account of symptoms, including flushing and diarrhoea, consistent with the carcinoid syndrome, and the suspicion of a gastrointestinal tumour, a plasma gut hormone panel was additionally requested following cessation of omeprazole therapy, and urinary 5-HIAA excretion was assessed (Table 1).
Results of gut hormone and urinary 5-HIAA biochemistry profile
5-HIAA, 5-hydroxyindoleacetic acid
Results of the gut hormone profile demonstrated elevated plasma chromogranin A and B, and elevated 24-h urinary 5-HIAA excretion, consistent with a serotonin-secreting GEP-ET. Immunohistochemistry of the patient's liver biopsy demonstrated positive immunostaining for CAM5.2, synaptophysin and chromogranin A, confirming the diagnosis of a metastatic, well-differentiated, gastroenteropancreatic endocrine carcinoma, with a Ki67 proliferation index of 15–20%. This was subsequently confirmed by a second centre (Figure 1).
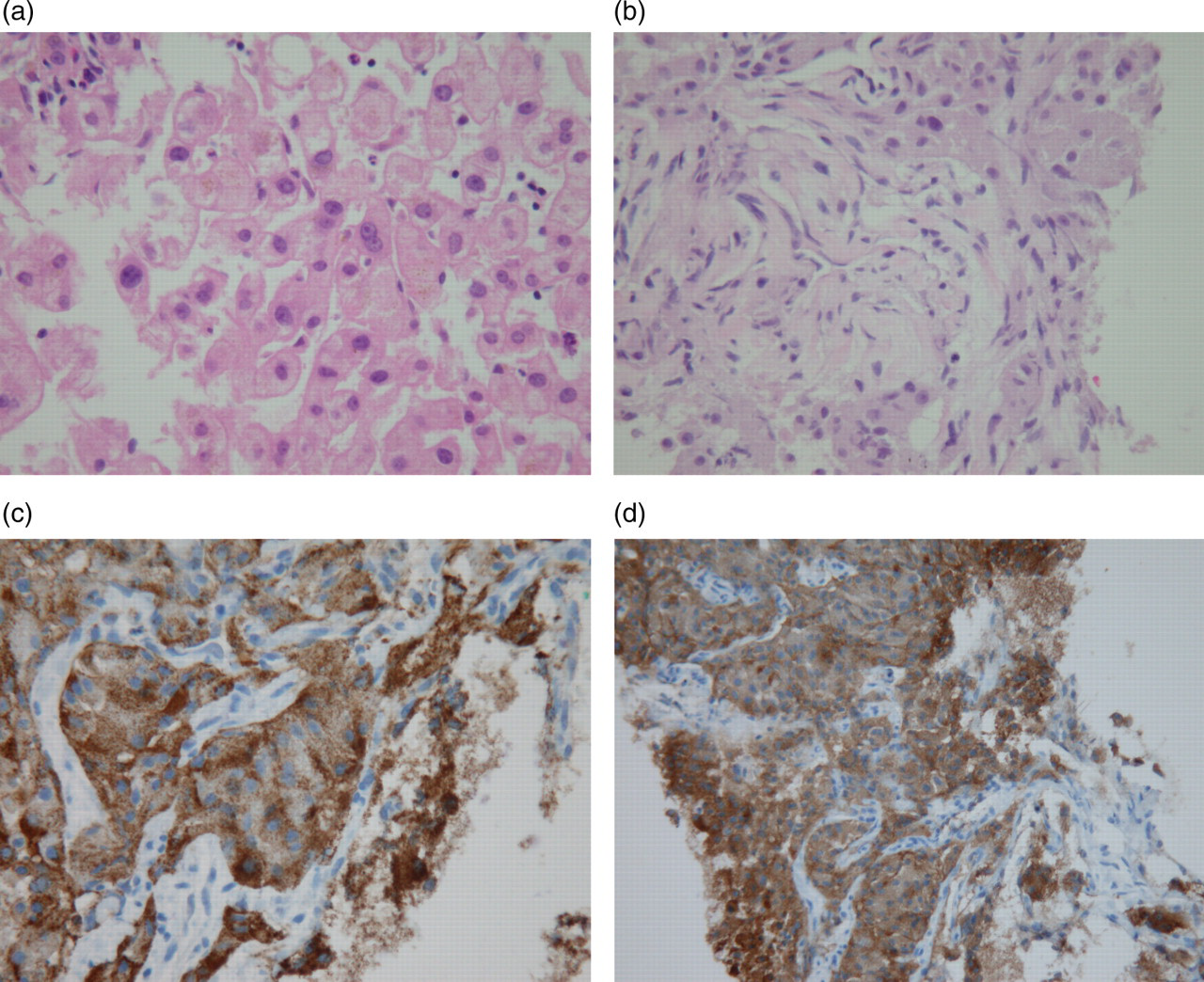
Haematoxylin/eosin-stained biopsy of (a) a portion of this patient's non-diseased liver, (b–d) portions of this patient's metastatic liver tumour. Biopsy tissue in (c) and (d) exhibit immunoreactivity for chromogranin A and synaptophysin, respectively, both specific markers of neuroendocrine cells
In the following month, the patient's hypoglycaemic symptoms increased in frequency and severity, requiring ingestion of glucose drinks every three hours during the night to relieve symptoms. Although the patient's LFTs had deteriorated considerably – ALP 476 U/L (30–130), gamma glutamyl transferase 1206 U/L (1–71), ALT 298 U/L (1–41), albumin 40 g/L (35–50) and total bilirubin 21 μmol/L (1–20) – the severity of hypoglycaemia was thought to be inconsistent with the extent of hepatic tumour burden. Following a supervised three-hour fast, plasma insulin and C-peptide concentrations were inappropriately elevated during hypoglycaemia, consistent with the diagnosis of an insulinoma – insulin 250.7 pmol/L, fasting glucose 2.2 mmol/L and C-peptide 2894 pmol/L.
Case follow-up
On diagnosis, the patient commenced treatment with diazoxide and SC Sandostatin to control hyperinsulinaemia-associated hypoglycaemia, also requiring administration of intravenous dextrose every three hours to prevent acute hypoglycaemic symptoms. The patient's clinical status deteriorated to World Health Organization (WHO) performance status 3, and was subsequently transferred to a specialist tertiary unit as an acute admission. On stabilization, the patient commenced Sandostatin LAR 30 mg monthly.
The patient underwent extensive radiological investigation: a CT scan identified a significant pancreatic mass indicating the primary tumour site and an octreotide scan demonstrated uptake in the liver, right femoral neck and in the body of the L2 vertebrae, but no secondary gastrointestinal (GI) lesion was observed. Widespread bone metastases primarily in the spine, pelvis and ribs were observed on a technetium-99m hydroxymethylene diphosphonate bone scan. On account of the hepatic tumour biopsy Ki67 proliferation index score, the patient commenced chemotherapy. Due to poor renal and liver function, standard cisplatin/etoposide and streptolysin/doxorubicin chemotherapies used for pancreatic tumours could not be used. Hence, the patient commenced with a chemotherapy regimen consisting of 12 cycles of oxaliplatin 85 mg/m2 d1 (14-day cycle) and 5-fluorouracil 2400 mg/m2 (over 2 days) with leukovorin.
During the course of chemotherapy, the patient's status improved clinically, biochemically and radiologically. Following the first cycle of chemotherapy, the patient's WHO performance status improved from 3 to 1, allowing further chemotherapy to occur on an outpatient basis. This improved clinical status was maintained throughout the entire course of chemotherapy during which the patient was treated with Sandostatin LAR without the need for any further dextrose infusions. In this time, the patient did not experience any hypoglycaemic episodes. Biochemistry results improved significantly; there was a marked improvement in LFTs and a significant sustained fall in CA19–9 reflecting improved liver and pancreatic function and alleviation of cholestasis. After the final course of chemotherapy, plasma chromogranin A concentration had fallen to 102 pmol/L, having reduced from the initial concentration of >1000 pmol/L (Table 2). The patient was not monitored for urinary 5-HIAA excretion as, by this point, symptoms attributable to the carcinoid syndrome were negligible and this was not considered the medical priority. Additionally, due to an inability to fast, plasma insulin levels could not be used to monitor PET response to chemotherapy. Following the final cycle of chemotherapy, response evaluation criteria in solid tumour (RECIST) measurements by a CT scan demonstrated a 12% reduction in hepatic and pancreatic tumour size compared with measurements taken mid-cycle, indicating stable disease.
Selected biochemistry results immediately before, during and following chemotherapy
ALP, alkaline phosphatase; AST, aspartate aminotransferase; GGT, gamma glutamyl transferase; LDH, lactate dehydrogenase; CA19-9, cancer antigen 19-9; CEA, carcinoembryonic antigen
The patient maintained WHO performance status 1 up to six months after the final cycle of chemotherapy. At this point, the patient began to report increasing lethargy and muscle aches, and experienced a hypoglycaemic episode. Biochemically, LFTs began to deteriorate (Table 2). At the time of writing this case report, the patient continued to receive active palliative treatment, being re-challenged with the chemotherapy regimen used previously.
Discussion
This report highlights a patient presenting with clinical syndromes associated with GEP-ET secretion of both insulin and serotonin. This may arise from the presence of two separate GEP-ETs each producing a different hormone, or a single tumour producing both hormones. The incidence of insulinoma (approximately 1/250,000 population/year) 4 and serotonin-secreting GEP-ET (approximately 1/40,000 population/year) 5 are such that coincidental presentation with two genetically unrelated lesions would be unlikely. Hence, the underlying pathology in this patient would more likely be accounted for by a solitary primary PET secreting a rare combination of humoral factors, or an unusual case of MEN1.
GEP-ETs are well documented for their ability to secrete multiple active humoral factors including amines, polypeptides and prostaglandins. Although over 40 different secretory products from GEP-ETs have been identified, 6 secretion of both serotonin and insulin is extremely rare, being documented in just two prior case reports. One of these cases detailed a patient with a primary hepatic endocrine tumour proven to secrete both serotonin and insulin. The clinical course of the patient's disease changed over a number of years from a carcinoid syndrome to a hyperinsulinaemic hypoglycaemic syndrome, 7 a similar clinical picture to the case reported here, albeit over a longer period of time. In a separate case report, an ileal carcinoid tumour that had metastasized to the liver was proven to secrete serotonin, insulin, calcitonin and gastrin. 8 In neither case was an insulinoma nor islet cell carcinoma observed at postmortem. With regard to the patient detailed in this case study, the patient's liver biopsy stained negative for insulin and C-peptide by immunohistochemistry, although the scant hepatic tumour biopsy material may have been of insufficient quality. Throughout the course of the illness, the patient was not sufficiently clinically stable to undergo pancreatic biopsy.
Owing to their biological and morphological heterogeneity, GEP-ETs were originally classified according to embryological origin; whether derived from the foregut (bronchus, stomach, duodenum, upper jejunum and pancreas), midgut (small intestine, appendix, cecum and proximal colon) or hindgut (distal colon and rectum). 9 The liver inactivates many of the humoral factors liberated by foregut GEP-ETs; hence, the presence of the carcinoid syndrome implies presence of liver metastases, since the secretory products liberated from these hepatic lesions drain directly into the systemic circulation. 2 Foregut GEP-ETs are also capable of secreting excessive quantities of histamine, 10 which may explain the intense pruritis experienced in this patient.
MEN1 is an autosomal dominant disorder with an estimated prevalence of one in 30,000 people. 11 The condition is defined as the combined occurrence of at least two of the three principal MEN1-related endocrine tumours – parathyroid adenomas, pancreatic islet cell endocrine tumours and pituitary tumours. 12 The majority of these tumours are hormone producing. The MEN1 gene is a tumour suppressor gene located on chromosome 11q13, encoding the protein menin. Over 400 somatic and germline mutations have been identified in the MEN1 gene causing nonsense, frameshift and missense mutations. 13 The condition is most commonly a heritable familial disorder, defined as an index MEN1 case plus at least one first degree relative with one of the three tumour types. 12 Less often the condition can occur sporadically.
In a study of 220 cases of MEN1, including familial and sporadic cases, insulinomas and serotonin-secreting GEP-ETs occurred in 27% and 4% of these patients, respectively. 14 The association of serotonin-secreting GEP-ETs with MEN1 is well documented, having higher prevalence in patients with MEN1 than those without. The majority of MEN1-associated serotonin-secreting GEP-ETs are of foregut origin and malignant metastasis is common. In contrast, serotonin-secreting GEP-ETs of midgut origin are a rare occurrence in MEN1. 15 Primary hyperparathyroidism arising from parathyroid adenomas is the most common MEN1 disorder, occurring in almost 100% of MEN1 patients over 50 years of age. 12 The patient reported in this case did not have a parathyroid hormone test; however, during inpatient assessment, the serum calcium remained consistently within the reference range – adjusted calcium range 2.26–2.36 mmol/L (2.18–2.63). As described previously, there was no family history of neoplasia.
Conclusions
We report a rare case of a patient with a metastatic GEP-ET with biochemical evidence of insulin and serotonin secretion. The clinical course of this patient changed from underlying symptoms of intense pruritis and features of the carcinoid syndrome to a severe hyperinsulinaemic hypoglycaemic syndrome. Extensive radiological investigation revealed a solitary primary pancreatic tumour indicating the presence of a metastatic PET secreting multiple active humoral factors, this being the first reported case of a PET secreting both insulin and serotonin. A demonstrable reduction in pancreatic tumour size was observed during chemotherapy, which coincided with significantly reduced plasma chromogranin A concentrations, further supporting this view. Due to biochemical evidence of an insulinoma and the association of serotonin-secreting GEP-ETs with MEN1, MEN1 should also be considered in such cases. In this case, the patient did not warrant further investigation for MEN1 due to their advanced age coupled with no evidence of primary hyperparathyroidism, atypical presentation and benign family history.
The WHO published guidelines in 2000, re-defining the terminology of GEP-ETs. The term ‘carcinoid’, which up to this point had been used to describe the majority of GEP-ETs, was deemed inappropriate due to the high degree of morphological and biological heterogeneity demonstrated by these lesions. To avoid the confusion associated with classifying all such lesions as carcinoids, a term increasingly being utilized by clinicians to describe serotonin-secreting GEP-ETs causing the carcinoid syndrome, carcinoid was replaced with GEP-ET. Distinction was made between well-differentiated endocrine tumours that exhibit benign behaviour or uncertain malignant potential, well-differentiated endocrine carcinomas characterized by low-grade malignancy and poorly differentiated endocrine carcinomas of high-grade malignancy. Furthermore, morphological and biological classification defined GEP-ETs by tissue localization, hormonal secretion and association with clinical syndromes. 9 Although the medical community has been slow to adapt to these recommendations, this point has been reinforced since. 16
Similarly, as our understanding of the genetic basis of the disease becomes more complex, the consensus definition of MEN1 (combined occurrence of at least two of the three principal MEN1-related endocrine tumours) appears limited. Serotonin-secreting GEP-ETs have a known association with MEN1 despite arising throughout the GI tract. Additionally, loss of heterozygosity of the 11q13 gene responsible for MEN1 has also been demonstrated in up to 50% of sporadic GEP-ETs of foregut origin, supporting a role for MEN1 gene inactivation in the development of these tumours. In contrast, losses of chromosomal arms 18p and/or 18q and 9p have been demonstrated in the development of midgut GEP-ETs, 17 emphasizing the distinction at the molecular level between GEP-ETs of differing embryological origin.
This case exemplifies these points. Awareness regarding the biological diversity of GEP-ETs, their secretory products and resultant clinical syndromes is crucial for effective investigation and diagnosis of such patients.
DECLARATIONS