
Abstract
We report a six-week-old boy with genitourinary structural abnormalities who presented with profound hyponatraemia and hyperkalaemia due to transient type 1 pseudohypoaldosteronism (PHA) precipitated by a urinary tract infection (UTI), which responded rapidly to intravenous saline and antibiotics. In infants with structural abnormalities of the urinary tract, we wish to highlight the importance of recognizing type 1 PHA and its association with a UTI since prompt and appropriate treatment rapidly corrects the associated metabolic abnormalities. Conversely, the identification of type 1 PHA in an infant should precipitate a search for a UTI and structural abnormalities of the urinary tract.
Introduction
Severe hyponatraemia and hyperkalaemia is a rare medical emergency in infancy and usually points to adrenal failure, typically congenital adrenal hyperplasia (CAH). The presence of urinary tract malformations, however, should alert to the possibility of type 1 pseudohypoaldosteronism (PHA), particularly in the presence of a urinary tract infection (UTI). 1–6
It is important to recognize transient type 1 PHA since it responds rapidly to specific treatment of the precipitating condition such as antibiotics for a UTI and aggressive fluid resuscitation with normal saline. We report a six-week-old boy with structural abnormalities of the urinary tract, who presented with transient type 1 PHA precipitated by a UTI.
Case report
A six-week-old boy known to have true chimerism of XX/XY and a mild obstructive uropathy presented as an emergency with a two-week history of worsening vomiting, weight loss and passing foul-smelling urine. He had a mild tachycardia of 138/min, was afebrile and did not look particularly unwell. A catheter specimen of urine (CSU) was cloudy and smelly, positive on dipstix for leukocytes, but negative for nitrites. Urgent microscopy per high power field identified >20 leukocytes and +++ microorganisms. Blood tests showed profound hyponatraemia and hyperkalaemia (Table 1). He was started on intravenous co-amoxiclav, a saline infusion and hydrocortisone to cover for the possibility of CAH. Hydrocortisone was stopped when his pre-treatment serum cortisol excluded adrenal failure (Table 1).
Clinical biochemistry results from presentation to resolution
A fully sensitive Escherichia coli was subsequently grown from the CSU. His electrolytes rapidly improved (Table 1) and he was started on oral sodium supplements. Subsequent urine cultures were negative. He was discharged home on oral sodium supplements and prophylactic trimethoprim. Other investigations on admission showed a very high serum aldosterone and plasma renin activity, but normal 17-hydroxyprogesterone (17OHP; Table 1).
One week after discharge, his serum electrolytes and plasma aldosterone were normal (Table 1) and therefore he was slowly weaned off oral sodium supplements. He remained well on prophylactic antibiotics that were stopped following successful surgery for first stage masculinization and correction of the urological malformation. He has since had an E. coli UTI, which was not associated with type 1 PHA and responded to antibiotics.
Although not confirmed by unavailability of past records at the time of the emergency admission, the child was born by elective caesarean section at 38 weeks gestation to consanguineous parents and was noted to have ambiguous genitalia at birth. Investigations excluded CAH, but the child was found to have true chimerism with 12/23 cells examined having a 46XX chromosome complement and the remaining 11/23 cells having a 46XY complement. Imaging studies demonstrated a hydrocolpos filling via a urogenital sinus and a mild right hydroureteronephrosis.
Discussion
Aldosterone, which is regulated by the renin–angiotensin system, increases renal distal tubular reabsorption of sodium and water at the expense of potassium. Deficiency of aldosterone (hypoaldosteronism) or resistance to its action (type 1 PHA), therefore, results in a similar biochemical picture of hyponatraemia, hypovolaemia, hyperkalaemia and metabolic acidosis, which although relatively rare in infancy is an important medical emergency. 5
The commonest cause of hypoaldosteronism is adrenal failure due to CAH as a result of 21-hydroxylase deficiency. The initial management of these children involves the collection of appropriate blood samples for cortisol, 17OHP, aldosterone and renin. The child is then immediately resuscitated by correcting the hypovolaemia, electrolyte abnormalities and presumed cortisol deficiency with a saline infusion and hydrocortisone as an IV bolus followed by an infusion. As soon as the initial treatment is started, the cause of the presumed adrenal crisis should be sought and treated. If CAH is confirmed by low cortisol and high 17OHP, management is then directed towards CAH. 7 If CAH is excluded, hydrocortisone is stopped, and then serum aldosterone will differentiate isolated hypoaldosteronism from type 1 PHA (Figure 1). The management of type 1 PHA then involves continuing salt and water replacement, treatment of the precipitating cause (e.g. antibiotics for an UTI) and exclusion of urinary tract structural abnormalities. An ultrasound of the urinary tract is recommended in all children under six months with an atypical UTI 8 irrespective of type 1 PHA. We also, however, suggest that any child over the age of six months with a UTI and type 1 PHA should have imaging studies of the urinary tract.
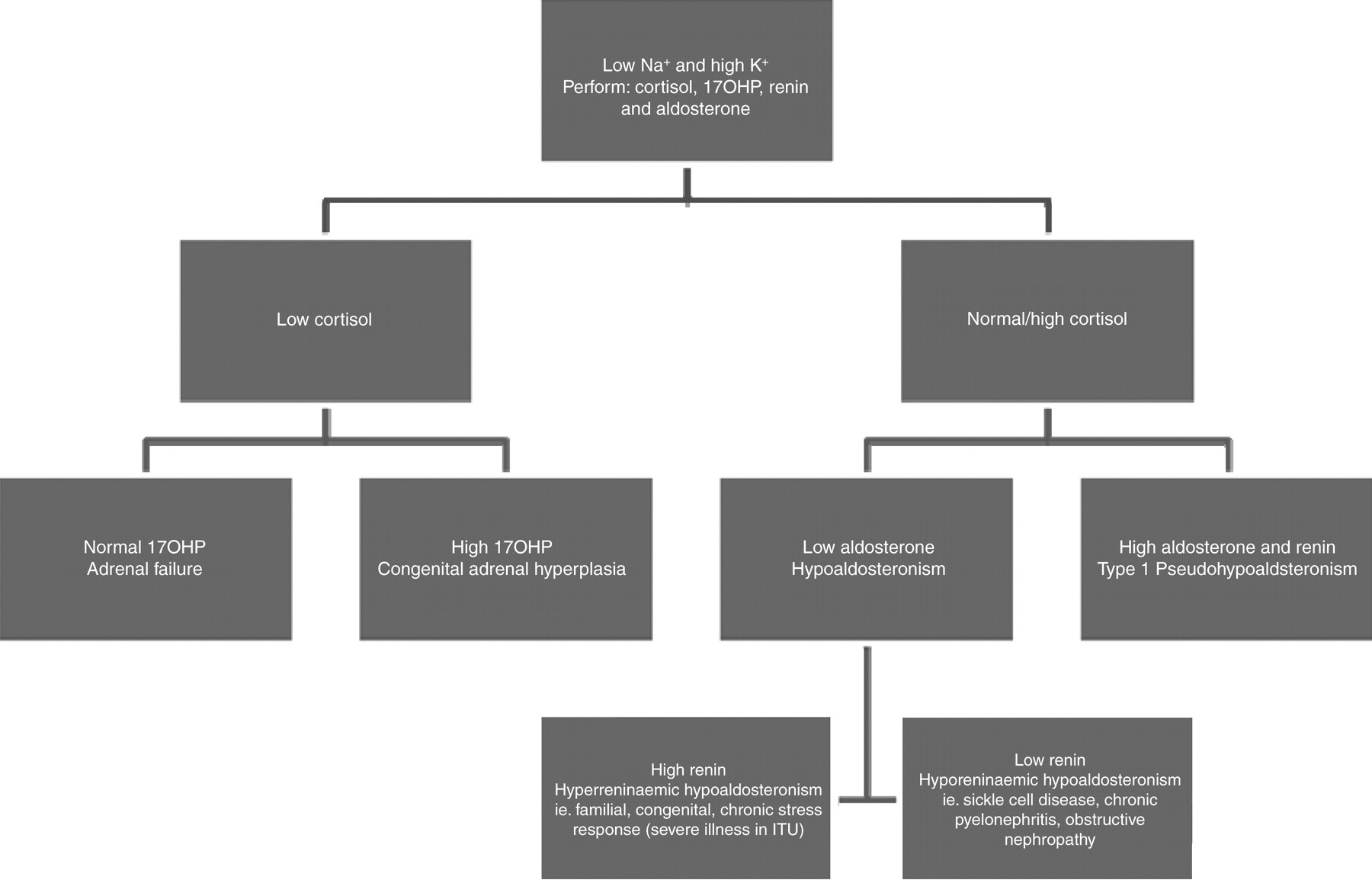
Differential diagnosis of hyponatraemia and hyperkalaemia. 17OHP, 17-hydroxyprogesterone; ITU, intensive care unit
In this particular case, CAH and adrenal failure were excluded by the appropriate serum cortisol and normal 17OHP (Table 1). The high aldosterone supported by the high renin activity was diagnostic of type 1 PHA (Table 1). PHA refers to a state of renal tubular resistance or unresponsiveness to the action of aldosterone characteristic of a heterogeneous group of disorders of electrolyte metabolism. Type 1 PHA may either be primary or secondary. Primary type 1 PHA is due to mutations of the mineralocorticoid receptor or epithelial sodium channel leading to renal or generalized resistance to aldosterone. 9 Type 1 PHA may also be secondary to disease or drugs affecting the kidneys such as nephritis, amyloidosis, angiotensin converting enzyme inhibitors or beta-blockers. 10 Although reported, it is not widely appreciated that secondary type 1 PHA may be transient in nature as exemplified in this case. It has been postulated that the presence of urinary tract abnormalities or infections or most commonly both predispose to the development of type 1 PHA; 5,6 however, the underlying pathology leading to mineralocorticoid resistance remains unknown. This case report, however, supports the notion that structural urinary tract abnormalities may be a necessary predisposition to the development of PHA since an E. coli UTI that occurred six months later did not trigger type 1 PHA once the urological abnormalities had been surgically corrected.
Conclusion
The acute clinical and biochemical presentation of transient type 1 PHA is identical to adrenal failure and may easily be confused with CAH, but these may be differentiated by careful interpretation of cortisol, renin and aldosterone results. Despite this, the initial management of both is similar, with transient type 1 PHA responding rapidly to treatment of the precipitating cause and replacement with salt and water emphasizing its transitory nature. If the child is not known to have structural abnormalities of the urinary tract, further diagnostic imaging of the genitourinary tract is indicated.
DECLARATIONS