
Abstract
Background
Mevalonic acid (MVA) is synthesized at an early and rate-limiting step in the biosynthesis of cholesterol by the enzyme hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase, and is a useful measure of statin efficacy or treatment. A liquid chromatography-tandem mass spectrometry (LC-MS/MS) method for the measurement of serum MVA has been developed.
Methods
Following the in vitro conversion of MVA to mevalonic acid lactone (MVAL) in the serum, MVAL and a deuterated internal standard were extracted using an online solid-phase extraction procedure. Chromatographic separation was achieved using a Luna PFP column (Phenomenex), with enhanced selectivity and improved resolution for polar compounds. A gradient system was used, with mobile phase comprising methanol and water (5 mmol/L ammonium formate buffer, pH 2.5). Analysis was performed using an API 5000 tandem mass spectrometer (Applied Biosystems) in positive electrospray ionization mode.
Results
The method showed excellent recoveries (98 ± 8%) and imprecision (intra-assay coefficient of variation of 2.2% [6.5 ng/mL] and 2.6% [10.5 ng/mL], and inter-assay coefficient of variation of 9% [10.5 ng/mL]). The assay provides a calibration range up to 50 ng/mL with a limit of detection at 0.1 ng/mL.
Conclusion
A simple, rapid and analytically specific method has been developed for the measurement of serum MVA, in the form of MVAL. The high analytical sensitivity of the method allows for accurate quantitation of MVAL in serum samples, both at the endogenous levels found in healthy individuals and in statin-treated patients where normal levels are expected to be greatly reduced through the inhibition of HMG-CoA reductase.
Introduction
The cholesterol biosynthetic pathway (Figure 1) begins in the cytosol with the formation of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) from acetyl-CoA (coenzyme A), amino acids and fatty acids, by the enzyme HMG-CoA synthase. 1 In a second, rate-limiting step, HMG-CoA is reduced by the microsomal enzyme HMG-CoA reductase, forming mevalonic acid (MVA). It is at this stage in the pathway that statins act as HMG-CoA reductase inhibitors to reduce MVA biosynthesis, and ultimately the circulating concentrations of cholesterol. 2 There is also evidence to suggest that exogenous cholesterol inhibits its own synthesis through negative feedback at the point of MVA production. 3
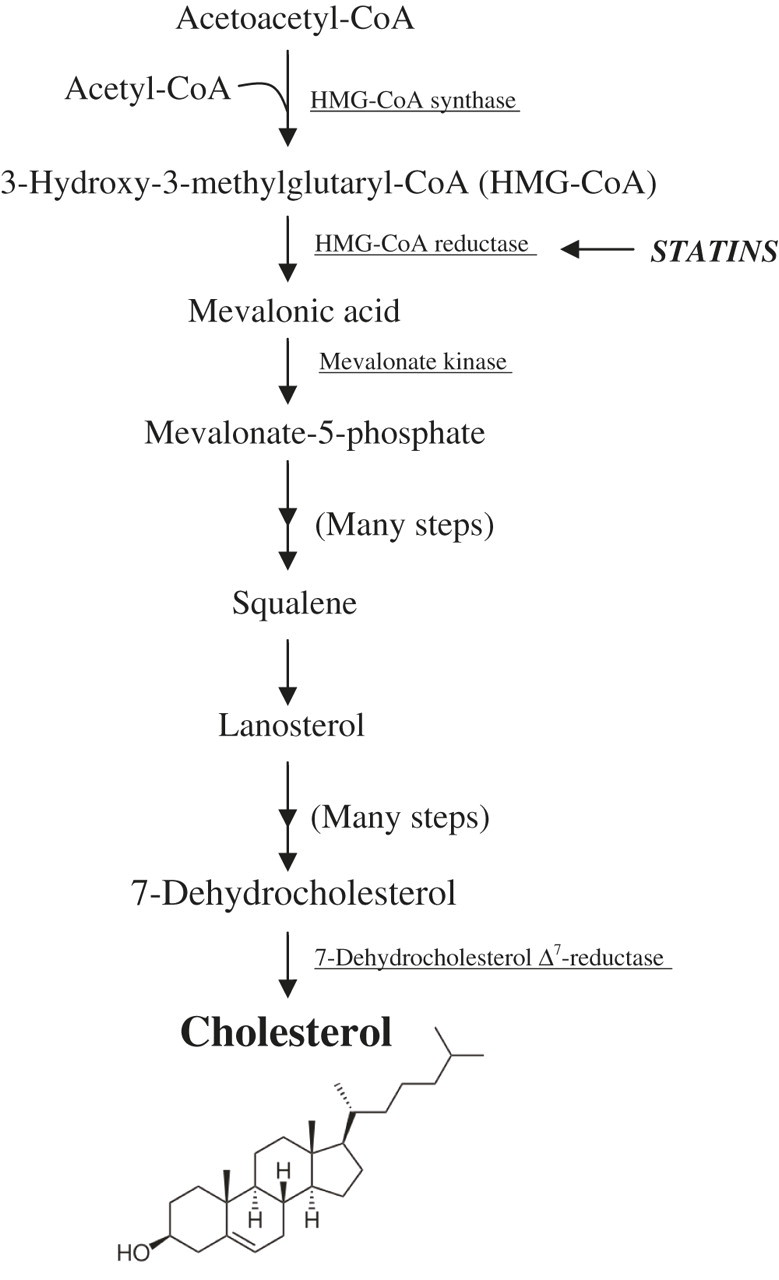
Biosynthesis of cholesterol
The plasma concentrations of MVA have been shown to correlate with the activity of hepatic HMG-CoA reductase in both rats and humans, and are related to the rate of cholesterol biosynthesis. 4 This suggests that MVA can be used as a good indicator of HMG-CoA reductase inhibition by statins and thus as a marker of statin activity. 5 Serum MVA levels could be measured in patients undergoing statin treatment as a more direct measure of statin efficacy than cholesterol, since MVA is synthesized at a much earlier, rate-limiting step in the biosynthetic pathway, and it is the step in which HMG-CoA reductase directly exerts its inhibitory effects. This could lead to a better understanding of the pharmacodynamics of statins, particularly with respect to any interindividual variation in response to statin therapy, and may consequently allow for improvements in treatment regimes and patient management.
Serum MVA might provide a useful measure of statin efficacy in clinical research trials such as those investigating the true effect of statins on vitamin D metabolism. In line with the theoretical considerations that cholesterol and vitamin D share 7-dehydrocholesterol (7-DHC) as a precursor in their biosynthetic pathways 1 (Figure 1), it would be predicted that for patients on statin therapy, the HMG-CoA reductase inhibitory effect of statins would result in not only a reduction in their circulating levels of cholesterol but also of vitamin D.
A wide variety of plasma and urine methods have been published for the quantitation of MVA, involving radioenzymatic assays, 6,7 immunoassays, 8 gas chromatography 3 (GC) and gas chromatography-mass spectrometry 9,10 (GC-MS). Until recently, most methods have employed GC, often involving the detection of a derivatized form of MVA or mevalonic acid lactone (MVAL). 2 The disadvantages of using GC-MS techniques mainly revolve around extensive sample preparation and the expense of equipment. Small bench-top GC-MS can be used, which does not require highly technical and expensive apparatus, although it is not accurate for serum or plasma analysis.
Very few liquid chromatography-tandem mass spectrometry (LC-MS/MS) methods have been reported. 2,4,11 Although GC provides significantly higher resolution, high-performance liquid chromatography (HPLC) methods can be considered superior to GC-based methods in terms of sample preparation, operation and high sample throughput. HPLC-MS/MS is increasingly being used in clinical laboratories owing to its increased analytical specificity and sensitivity for a wide range of analytes. The challenges associated with the development of serum or plasma-based methods for MVA are based around difficulties with extraction methods, and specificity and sensitivity of detection, since MVA is a polar, endogenous molecule present at nanogram levels in the blood. 2 MVA is of low molecular weight and the mass transitions used in MS detection are often a non-specific loss, thus allowing for greater interference from serum or plasma components on detection. 4 MS/MS provides a distinct advantage over conventional MS detection due to a phenomenon known as multiple reaction monitoring (MRM). MRM allows several mass transitions to be detected and quantified simultaneously, thus greatly enhancing analytical specificity, particularly for low molecular weight analytes such as MVA.
MVA exists in two forms, depending on the pH conditions of the surrounding environment. MVAL is obtained by the dehydration of MVA and turns back into MVA in water (Figure 2). MVA is converted to MVAL under acidic conditions, and MVAL can be converted back to MVA under basic conditions. Quantitation often initially involves this pH-dependent conversion of MVA to MVAL. This is generally followed by a solid-phase extraction (SPE) process prior to analysis of the extract, together with a deuterated MVAL internal standard. MVA can then be analysed by HPLC-MS/MS with an electrospray ionization (ESI) source, which is a sensitive detection method for polar molecules. 12 ESI can be set to positive mode for the detection of MVAL, 4 or to negative mode for MVA detection following the conversion of MVAL back to MVA. 2,11 The method reported in this paper provides a rapid, simple, analytically specific and sensitive HPLC-MS/MS method for the measurement of serum MVAL, using online SPE for sample clean-up and positive mode ESI for MS/MS detection. This method achieves recoveries superior to those previously reported in the literature, without the requirement for complex column-switching procedures. 4
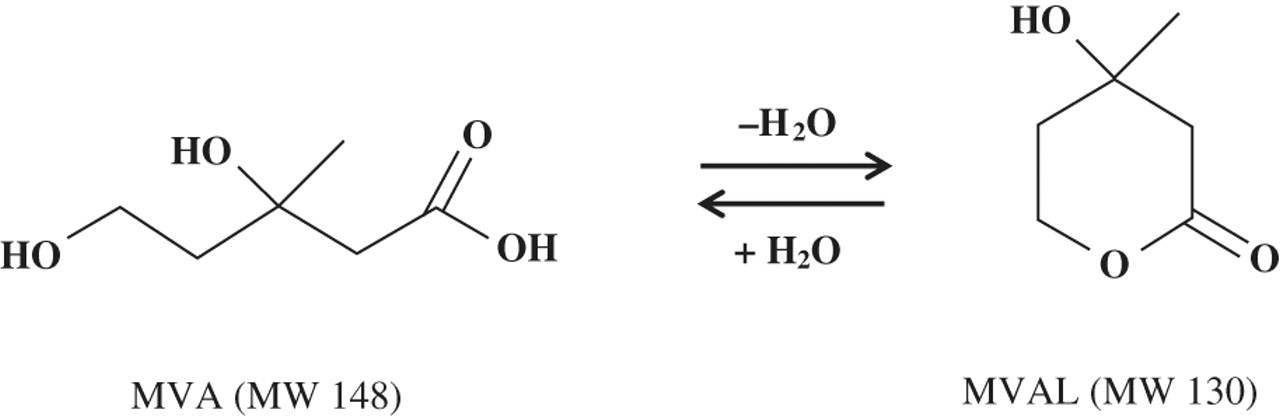
Chemical structures of mevalonic acid (MVA) and mevalonic acid lactone (MVAL)
Materials and experimental procedures
Chemicals and reagents
Commercial MVAL (97%) was obtained from Sigma-Aldrich (Poole, UK) and an internal standard, deuterated MVAL (D4-MVAL) from CDN isotopes (Pointe-Claire, Quebec, Canada). A Luna PFP (3 μm, 2.0 × 150 mm) column and Strata-X-CW (25 μm, 2.0 × 20 mm) online extraction cartridge for online SPE were purchased from Phenomenex (Macclesfield, UK). All solvents and other reagents were of analytical grade (VWR International, Lutterworth, UK). Control human serum for method development and preparation of quality control samples was obtained from human volunteers in the directorate of laboratory medicine at Birmingham Heartlands Hospital, UK. Serum from statin-treated patients for method validation was obtained from lipid clinics at Birmingham Heartlands Hospital, UK. C.f.a.s. (calibrator for automated systems) calibrator material for spiked control samples was purchased from Roche diagnostics (West Sussex, UK).
HPLC-MS/MS instrumentation
Analysis was performed using a Prominence ultra-fast liquid chromatography (HPLC) system (Shimadzu Scientific Instruments, Milton Keynes, UK), with a two-position switching valve (Valco Instruments Co Inc, Houston, TX, USA) for online extraction. The HPLC system was coupled to an API 5000 tandem mass spectrometer with a QJet ion guide (Applied Biosystems, Warrington, UK) using a turbo ESI source. The software package used was Analyst version 1.5 (Applied Biosystems).
Other apparatus
Sample extracts were mixed using a Grant Bio variable speed vortex mixer (Fisher Scientific UK Ltd, Loughborough, UK) and centrifuged using an MSE Centaur microcentrifuge (VWR International).
HPLC-MS/MS conditions
Chromatographic separation was achieved using a Phenomenex Luna PFP column, which enables enhanced selectivity and improved resolution for polar compounds. Online sample extraction was carried out using a pre-column Strata-X-CW online extraction cartridge, which was connected to a Valco switching valve. The column oven was maintained at a temperature of 40°C throughout experimental procedures. A gradient method (as detailed in Table 1) was used, with mobile phase comprising (A) water (5 mmol/L ammonium formate, adjusted to pH 2.5 with formic acid) and (B) methanol (5 mmol/L ammonium formate, adjusted to pH 2.5 with formic acid). An injection volume of 100 μL and a flow rate of 0.3 mL/min were used, with a total run time of 6.5 min. Ionization was achieved in a positive mode ESI for the measurement of MVA in the form of MVAL. The MS/MS parameters are detailed in Table 2. MRM was used with MVAL reporter transitions at m/z 131/69 and 131/43, and a transition at m/z 135.3/74.9 for the internal standard (D4-MVAL).
High-performance liquid chromatography gradient programme
MS/MS parameters for MVAL and D4-MVAL analysis
DP, declustering potential; EP, entrance potential; CXP, collision cell exit potential; CE, collision energy; CAD, collisionally activated dissociation gas; GS1, source nebulizer gas; GS2, source auxiliary gas; TEM, source temperature; CG, curtain gas; IS, ion spray voltage; MS/MS, tandem mass spectrometry; MVAL, mevalonic acid lactone
Calibration standards and controls
A standard stock solution of MVAL was prepared in methanol to a concentration of 1 g/mL, and a working solution of MVAL at 50 ng/mL was prepared by diluting the standard stock solution in water. Calibration standards at five different concentrations (ranging between 0.5 and 50 ng/mL) were prepared by serial dilution of the MVAL working solution in water. A deuterated internal standard (D4-MVAL) stock solution was prepared in methanol to a concentration of 0.05 g/mL. A working internal standard was prepared by dilution of the stock solution in water to a final concentration of 500 ng/mL.
A low-quality control (LQC) sample was prepared to a target concentration of 1 ng/mL by spiking MVAL into water. A high-quality control (HQC) sample was prepared to a target concentration of 10 ng/mL by spiking the appropriate amount of MVAL into a pool of C.f.a.s. calibrator material (lyophilized calibrator based on human serum, containing normal concentrations of several common analytes) following an initial assessment of the endogenous levels of MVAL present. A third quality control sample was also prepared from a pool of human serum and analysed for its endogenous MVAL concentration.
Sample preparation
Samples were prepared via the addition of 10 μL of working internal standard to 200 μL serum in tecan tubes. This was followed by the addition of 100 μL sulphosalicylic acid for protein precipitation and the pH-dependent conversion of MVA to MVAL. Sample preparations were thoroughly mixed and centrifuged at 13,000 rpm for five minutes. The resulting supernatants were then transferred into glass vials for direct injection into the HPLC-MS/MS system.
Method validation
Recovery
The accuracy of the method was assessed through a recovery experiment. Serum samples (n = 5) were spiked with MVAL at 200 ng/mL and baseline samples were spiked with water. Spike volumes were minimized to ensure a dilution of less than 10%. Samples were subjected to analysis by the final extraction and HPLC-MS/MS protocol. Recovery was then calculated, taking into consideration the dilution of the ‘amount added’ in the total sample mixture.
Imprecision
The analytical imprecision of the assay was determined via the calculation of coefficient of variation (%CV) for intra- and inter-assay precision experiments. The intra-assay precision was determined through the analysis of patient serum samples at concentrations of 6.5 and 10.5 ng/mL (n = 11). The inter-assay precision was determined by the analysis of five batches of five serum sample aliquots (at 10.5 ng/mL) on five consecutive days.
Analytical range
The analytical range was determined through the analysis of a series of MVAL reference solutions in water, ranging from 3.125 to 100 ng/mL.
Limit of detection
The limit of detection was determined through the analysis of MVAL solutions at 10, 1 and 0.1 ng/mL (in water) to identify the lowest concentration at which the signal:noise (S/N) ratio was >3. The S/N ratio was calculated from Gaussian smoothed data using the S/N script tool (Applied Biosystems).
Ion suppression
An ion suppression experiment was carried out to determine the matrix effect of compounds in the serum extracts. A standard solution of MVAL (50 ng/mL in water) was infused directly into the mass spectrometer at 10 μL/min using a syringe driver and an extracted serum sample was injected into the HPLC system simultaneously. The effect on the signal intensity was observed over the time course of the assay.
Stability study
The stability of MVA in serum was tested at 4°C after a one-week period, at −20°C after three months and at −80°C after six months (n = 5). Statistical analysis was carried out via one-way analysis of variance, using Analyse-it for Microsoft Excel software.
Determination of a reference range
Following full method validation, 54 serum samples obtained from healthy human volunteers were analysed for their endogenous levels of MVAL. LQC and HQC samples were also analysed to ensure analytical accuracy of sample measurement. All volunteers were statin-naïve, and the sample group encompassed a 31% male and 81% Caucasian (15% Asian, 4% Afro-caribbean) population. A reference range was determined via non-parametric statistical analysis using Analyse-it for Microsoft Excel software.
Clinical application of serum MVAL
The validated method was applied to quantify serum MVAL concentrations in a group of 15 hyperlipidaemic outpatients attending lipid clinics at Birmingham Heartlands Hospital. This group encompassed a 40% male population with a mean age of 58 (±7). Diagnoses included hypercholesterolaemia (27%), possible familial hypercholesterolaemia (40%), post-menopausal hypercholesterolaemia (13%), type III hyperlipidaemia (7%) and metabolic syndrome (13%). Patients were prescribed a statin in the form of simvastatin (47%), atorvastatin (20%) or rosuvastatin (33%), as part of their routine care plan. To determine the effect of statins on serum MVAL levels, MVAL was measured in these patients at baseline (when statin-naïve) and at three months post-commencement of their statin therapy. Statistical analysis was carried out via a Wilcoxon matched-pairs signed-ranks test, using Analyse-it for Microsoft Excel software.
Results
HPLC-MS/MS method
Multiple reaction monitoring
MRM was utilized for the detection of two MVAL mass transitions and the D4-MVAL transition (as detailed in Table 2), thus providing high analytical specificity for MVAL detection. The correct MVAL fragments were confirmed during the MS/MS tuning procedure by comparing the observed and expected fragmentation patterns using a fragment interpretation tool within the Analyst software (Applied Biosystems). Figure 3 illustrates the product ion mass spectrums produced for MVAL and D4-MVAL during tuning. The [M]+ ion at m/z 131 was identified as the precursor ion for MVAL, and the most abundant product ions selected for MRM optimization were at m/z 43 and 69. The precursor ion for D4-MVAL was identified as the [M]+ ion at m/z 135.3 and the predominant product ions at m/z 74.9 and 61 were selected for optimization. Quantification was determined by the m/z 131/69 transition, and the second MVAL transition at m/z 131/43 was used to confirm the specificity of detection.
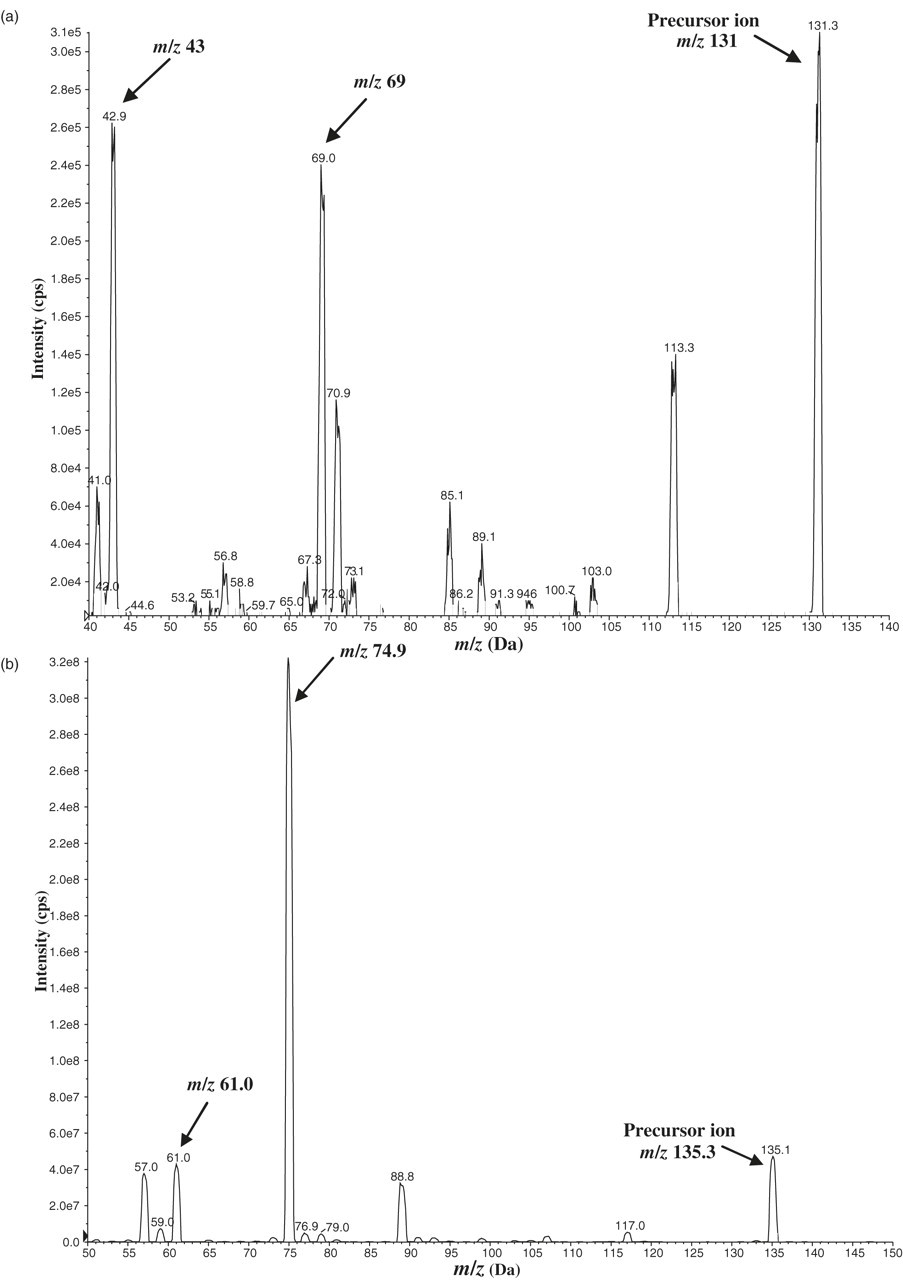
Product ion mass spectrum for (a) mevalonic acid lactone (MVAL) and (b) D4-MVAL (arrows indicate the precursor ions and most abundant product ions selected for optimization)
Patient sample analysis
Figure 4 illustrates a chromatogram of an extracted serum sample containing D4-MVAL internal standard. Excellent chromatography was observed, with sharp, symmetrical peaks for all three MRM transitions at a retention time of 4.8 min.
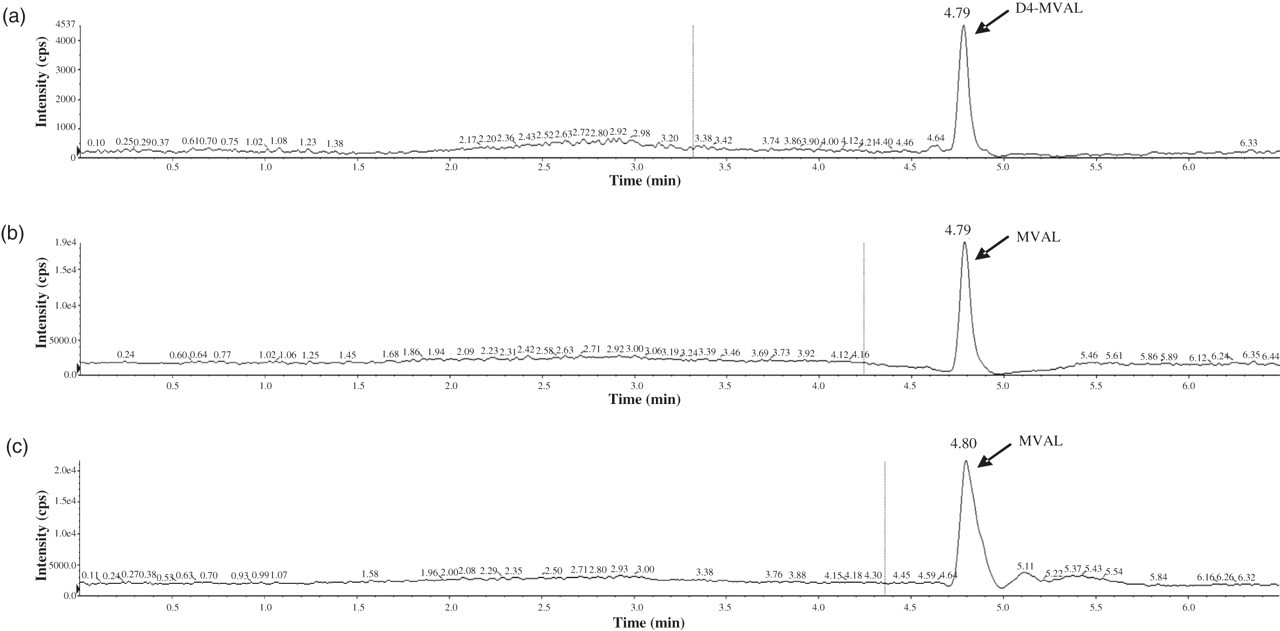
Chromatogram illustrating multiple reaction monitoring of an extracted patient serum sample: (a) D4-MVAL (mevalonic acid lactone) m/z 135.3/74.9, (b) MVAL m/z 131/69 and (c) MVAL m/z 131/43
Calibration standards and controls
The final calibration standards were prepared at concentrations of 0.5, 12.5, 25, 37.5 and 50 ng/mL. Figure 5 shows a typical standard curve with five calibration points, and a measuring range between 0.5 and 50 ng/mL.
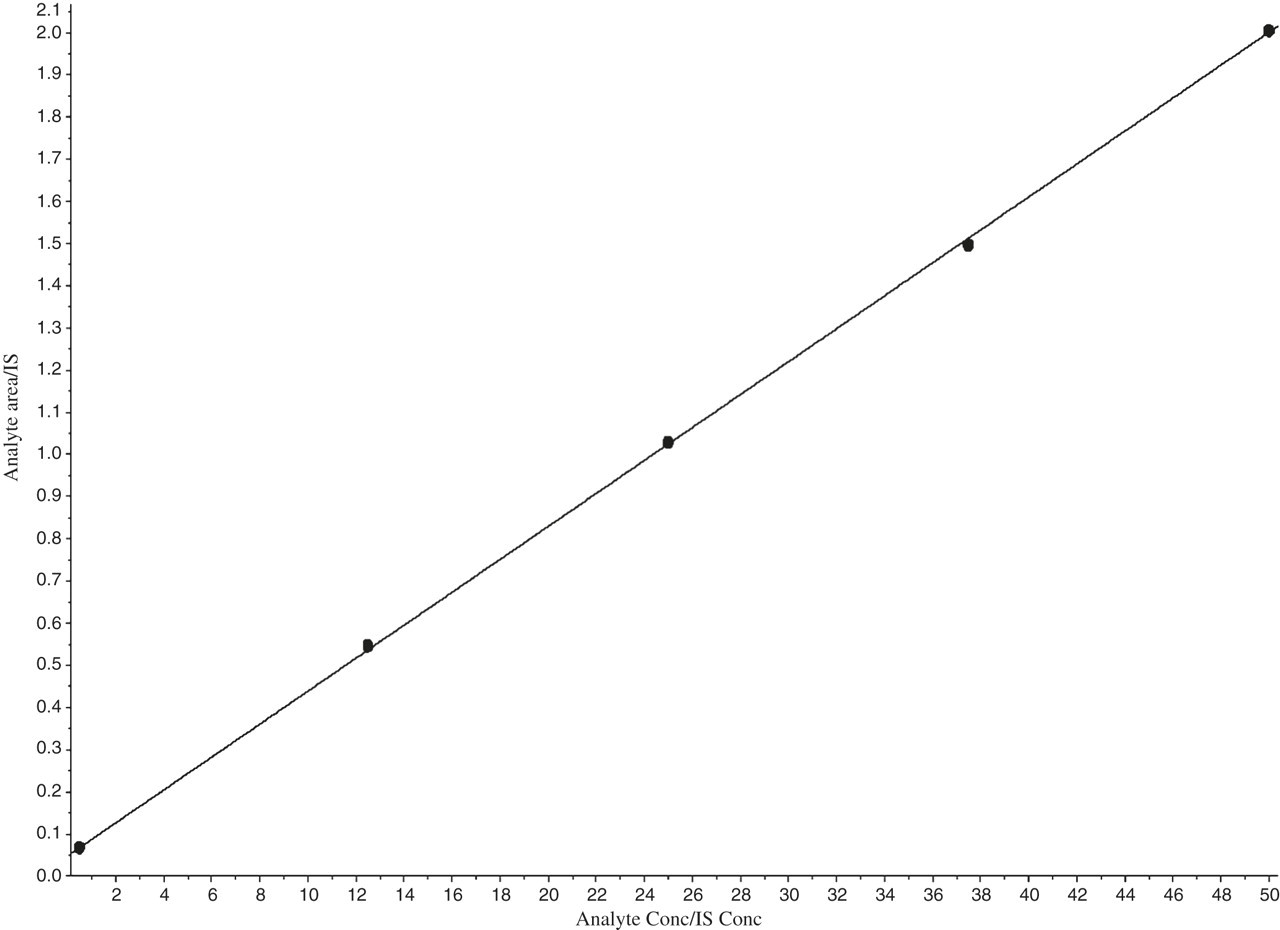
Calibration curve for mevalonic acid lactone, with a measuring range between 0.5 and 50 ng/mL. IS, internal standard
The endogenous level of MVAL in the C.f.a.s. calibrator material for use as an HQC material was measured at 2 ng/mL (n = 1). The mean concentrations measured in the spiked LQC and HQC samples were 2.8 ng/mL (n = 7) and 11.6 ng/mL (n = 5), respectively. The third quality control sample of pool of human serum had a mean concentration of 15.3 ng/mL (n = 6).
Method validation
The mean recovery for the serum extraction of MVAL was 98 ± 8% (n = 5). The intra-assay %CVs at concentrations of 6.5 and 10.5 ng/mL (n = 11) were 2.2% and 2.6%, respectively, and the inter-assay %CV at a concentration of 10.5 ng/mL (n = 5) was 9.0%. The assay provides a calibration range up to 50 ng/mL (R 2 = 0.9997) with a limit of detection at 0.1 ng/mL (S/N ratio >3). A low level of ion suppression was observed, and this was at a similar level for both MVAL mass transitions at approximately 4.8 min, where elution of the MVAL peak would be expected (Figure 6). Higher levels of ion suppression were observed in the regions between 2.5 and 3.0 min, and between 5.0 and 5.5 min of the assay run. Following statistical analysis, samples were found to be stable at 4°C after one week, with no significant difference (P > 0.05) in MVAL concentrations between these samples and those stored at −20°C immediately after blood sampling (−20°C samples were taken as a baseline value). Samples were also found to be stable at −20°C after a three-month period and at −80°C after six months, with no significant difference (P > 0.05) in MVAL concentrations between these samples and the – 20°C baseline samples.
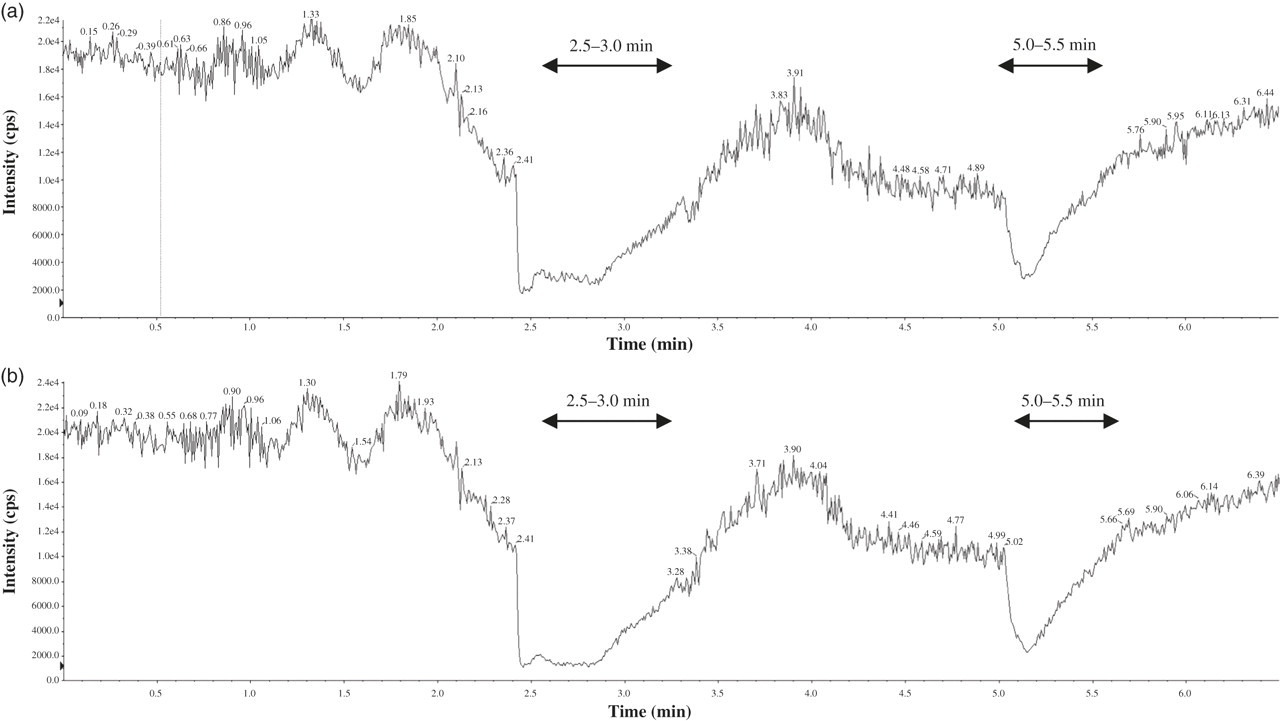
Ion suppression effect of a serum extract on the signal of an infused mevalonic acid lactone standard (a) m/z 131/69 and (b) m/z 131/43 (arrows indicate the regions of ion suppression observed)
Determination of a reference range
A reference range for MVAL was determined following analysis of endogenous MVAL levels in the human serum samples. An Anderson–Darling normality test confirmed a normal distribution of serum MVAL (P > 0.05) and parametric statistical analysis was used to determine a reference range of 4.8–18.2 ng/mL (mean MVAL concentration 10.7 ng/mL).
Clinical application of serum MVAL
Statin treatment produced a statistically significant decrease in serum MVAL (18.1 ± 7 versus 9.2 ± 3.4, P < 0.0001), which equated to a mean decrease of 49%. The reduced levels of serum MVAL translated into a statistically significant decrease in serum total cholesterol levels (7.2 ± 1.3 versus 5.3 ± 0.9, P < 0.05).
Discussion
A rapid, simple, analytically sensitive and specific method was required for the measurement of MVA in the serum, as a marker of statin efficacy in clinical trial patients undergoing statin therapy. Until recently, serum or plasma MVA has most commonly been measured by GC or GC-MS methodologies, involving complex sample extraction and derivatization steps. 3,9,10 Very few HPLC-MS/MS methods have been developed so far, despite the superiority of this technique in terms of analytical sensitivity and specificity of detection owing to its MRM capabilities.
Those HPLC-MS/MS methods previously reported in the literature have struggled to achieve sufficient analytical sensitivity for the detection of MVAL in positive ESI mode without the need for complex column-switching techniques, 4 and have failed to reach recoveries close to 100% (as detailed in Table 3). HPLC-MS/MS in negative ESI mode has previously been the favoured methodology for MVA detection. In two different studies, 2,11 it appears that a significantly better analyte response was achieved for the detection of the MVA anion in negative mode than that for the detection of MVAL in positive mode. However, the method reported here showed a much improved analytical sensitivity, in terms of peak height response, for the detection of MVAL in positive mode ESI, particularly when the method was transferred to a highly sensitive API 5000 (Applied Biosystems) MS/MS instrument. The MVAL mass transitions at m/z 131/69 and 131/43, and the D4-MVAL internal standard transition at m/z 135.3/74.9 were selected for the final method owing to their consistently greater signal intensity. Quantification was carried out using the 131/69 transition due to its greater sensitivity. It would be expected that the low product ion masses chosen as reporter ions for the final method would give rise to a high level of background noise. However, the optimized mass transitions provided adequate sensitivity of detection with a minimal level of background noise.
Accuracy and imprecision data for the current method and previous methods in the literature
ESI, electrospray ionization; HPLC, high-performance liquid chromatography; CV, coefficient of variation; MS/MS, tandem mass spectrometry
During HPLC method development, a wide variety of columns matrices, including the Luna HILIC (hydrophilic interaction liquid chromatography) and several candidate reversed phase columns, were trialled for MVAL separation. A much improved peak shape and retention was observed with the Luna PFP column (pentafluorophenyl propyl ligand bonded to a silica matrix), which enables enhanced selectivity and improved resolution for polar compounds under reversed phase conditions. Optimum peak shape and a retention time of 4.8 min were achieved using a gradient system (as detailed in Table 1), with mobile phase comprising methanol and water (5 mmol/L ammonium formate buffer, pH 2.5). This provided a rapid HPLC method, with a run time of 6.5 min per sample.
Liquid–liquid extraction was initially trialled for the extraction of MVAL from the serum, since this has historically been a favoured technique for chromatographic analyses, and it is a simple and cheap method. In this study liquid–liquid extraction experiments were unsuccessful, with poor extraction efficiencies being observed. SPE was subsequently trialled, since the wide variety of SPE sorbents available allow highly efficient extraction of specific analytes from a matrix of interfering compounds. 13 Several SPE cartridges were tested, with a Strata XC (reversed phase mode) cartridge most successfully demonstrating MVAL retention and extraction efficiency. However, this manual SPE technique required the use of dedicated extraction apparatus and was found to be particularly labour-intensive and time-consuming, with limited capacity in terms of sample batch size. Thus a sample preparation method simply using sulphosalicylic acid protein precipitation, followed by extraction using a pre-column Strata-X-CW online extraction cartridge, was developed to provide a rapid sample preparation method for up to 80 samples per run.
Adequate analytical sensitivity was important to enable accurate quantification of MVAL in human serum, both at the endogenous levels found in normal individuals, and in statin-treated patients where normal levels are expected to be greatly reduced through the inhibition of HMG-CoA reductase. The required sensitivity was achieved with an S/N ratio of >3 at 0.1 ng/mL, which is indicative of a low background signal and a clear definition between the analyte of interest and any co-extractants. It was necessary to prepare calibration standards and LQC material using water as a surrogate matrix to allow for a sub-ng/mL limit of quantification. The assay has a calibration range of 0.5–50 ng/mL, with the upper limit representing MVAL concentrations at least five-fold in excess of those found in healthy individuals, since the reported normal range in humans is 1.5–11.8 ng/mL. 2 A mean endogenous concentration of 8.47 ng/mL (range 6.85–9.96 ng/mL) has also been quoted in the literature. 4 The method described thus provides sufficient analytical range for clinical use.
The method was fully validated for the quantification of MVA in patient samples. Excellent recoveries (98 ± 8%) were achieved, and these results are far superior to previous methods where recoveries ranged between 21% and ≥80%. 2,4,11 It is important to note that the use of a deuterated internal standard, and subjecting the calibration standards and QC material to the same extraction procedure, corrects for recoveries of <100%. Good intra- and inter-assay %CVs were observed during imprecision experiments, which compare favourably with previous HPLC-MS/MS methods. Table 3 summarizes the accuracy and imprecision data for the method of the current study and previous methods. During ion suppression experiments, an insignificant level of ion suppression from serum extracts was observed in the region of the chromatogram where the MVAL peak elutes. Combined with near 100% recoveries in patient samples, this method thus appears to be free of significant interference by solutes and endogenous compounds from the serum matrix, further indicating a highly efficient sample extraction procedure. MVA was found to be stable in serum samples at 4°C for a one-week period, at −20°C for three months and at −80°C for six months, thus providing adequate stability for the storage and efficient analysis of clinical trial samples.
The validated method was successfully applied to quantify serum MVAL concentrations in a group of healthy, statin-naïve volunteers, to determine a mean endogenous level of 10.7 ng/mL and a reference range of 4.8–18.2 ng/mL. These figures are not dissimilar to those previously quoted in the literature, as described earlier. 2,4 The potential clinical utility of MVA as a marker of statin efficacy was also demonstrated through the measurement of serum MVAL in a group of statin-treated patients, both prior to and post-commencement of their treatment regimen. Marked reductions in serum MVAL concentrations (mean reduction 49%) were observed three months post-statin commencement. This reduction is consistent with the inhibition of the HMG-CoA reductase activity of statins, and supports the findings of another similar study previously reported in the literature. 7 A mean pre-treatment MVAL concentration of 18.1 ng/mL was identified in these patients, which is greatly increased when compared with the observed mean endogenous levels in healthy populations, and lies at the upper limit of the reference range determined in this study. This finding may have implications for the expected MVAL levels in patients with specific lipid disorders, such as familial hypercholesterolaemia, and suggests a requirement for further cross-sectional studies in these patient groups. As would be expected, the reduced levels of serum MVAL translated into a marked reduction in serum total cholesterol levels. The analytical method described is thus sufficient to measure the normal endogenous levels of serum MVAL as well as those in statin-treated patients.
Conclusions
A simple, rapid, analytically sensitive and specific HPLC-MS/MS (positive ESI mode) method has been developed for the measurement of serum MVA, in the form of MVAL. This method has been fully validated to allow accurate quantification of MVAL in serum samples. High analytical specificity is provided through an efficient online SPE procedure, combined with the MRM capabilities of HPLC-MS/MS technology. This method provides sufficient sensitivity of detection in the serum of both healthy individuals and patients undergoing statin therapy.
Patients are increasingly being prescribed statins as part of their routine health care, thus serum MVAL analysis might aid in the improvement of such pharmacotherapy and patient management. The measurement of serum MVAL may help in the identification of statin-intolerant patients, and might be used to determine whether hypercholesterolaemia is occurring as a result of the increased production of cholesterol, or the inability to remove cholesterol from the circulation by, for example, the LDL-receptor. In association with hypercholesterolaemia, high levels of serum MVA might suggest increased synthesis of cholesterol, whereas normal or low levels could be indicative of impaired removal. The patient's lipid profile may be interpreted taking into consideration the serum MVAL levels and thus direct more appropriate management. Serum MVAL might also be used as a direct measure of statin efficacy in clinical research trials such as those investigating the true effect of statins on vitamin D metabolism.
DECLARATIONS