
Abstract
We present four cases with clinical and biochemical hypocalcaemia and evidence supportive of hypoparathyroidism. One case had been previously ascribed a diagnosis of idiopathic hypoparathyroidism. Following the detection of relative hypercalciuria, all cases were found to have autosomal dominant hypocalcaemia with hypercalciuria and mutations of the calcium-sensing receptor gene, of which two were novel. Increased awareness of this condition and access to genotyping enables prompt accurate diagnosis and cascade screening of first-degree relatives.
Introduction
Familial hypoparathyroidism has been recognized since the 1950s with the first report of probable autosomal dominant transmission within an asymptomatic family in 1987. 1 In 1993, a loss of function mutation of the calcium-sensing receptor (CaSR) gene was shown to be associated with familial benign hypocalciuric hypercalcaemia, 2 and the following year a gain of function mutation (E127A) in this gene, causing a form of autosomal dominant hypocalcaemia, was identified. 3 Associated with relative hypercalciuria, this condition is now referred to as autosomal dominant hypocalcaemia with hypercalciuria (ADHH). It has become increasingly apparent that many cases previously ascribed the diagnosis of idiopathic hypoparathyroidism are actually due to activating mutations of the CaSR gene. With increased awareness of this condition and access to genotyping, the diagnosis is now made in a more timely manner with appropriate cascade screening of first-degree family members.
We report four cases of ADHH, one of which had been previously assigned a diagnosis of idiopathic hypoparathyroidism. Two novel mutations of the CaSR gene are described and a brief review of the literature presented.
Case reports
Case one
A nine-year-old girl presented acutely following a focal seizure associated with a febrile illness. A computed tomography head scan demonstrated extensive calcification in the basal ganglia. Serum corrected calcium was low at 1.63 mmol/L (2.2–2.6 mmol/L), phosphate elevated at 3.11 mmol/L (0.8–1.4 mmol/L) and magnesium just below the lower limit of the reference range at 0.67mmol/L (0.68–0.90 mmol/L). Creatinine was normal. 25-Hydroxyvitamin D (25[OH]D) was 82 nmol/L (37–190 nmol/L), 1,25-dihydroxyvitamin D (1,25[OH]2D) was 60 pmol/L (47–161 pmol/L) and parathyroid harmone (PTH) undetectable at <0.4 pmol/L (1.3–7.6 pmol/L). A spot urinary calcium/creatinine ratio was inappropriately elevated at 1.56 (<0.45) at a time when her corrected calcium was 1.97 mmol/L. There was no family history of hypo- or hypercalcaemia. She was commenced on calcium, magnesium and vitamin D supplementation and further investigations including renal tract imaging were unremarkable. She was subsequently reviewed at age 19 y and CaSR gene mutation analysis was performed. This confirmed her to be heterozygous for the previously described missense CaSR gene mutation c.386G > A (C129Y). 4
Case two
A 33-year-old woman with epilepsy presented with recurrent seizures. Investigations identified hypocalcaemia with serum corrected calcium at 1.9 mmol/L (2.2–2.6 mmol/L). Phosphate was mildly elevated at 1.5 mmol/L (0.8–1.4 mmol/L) with normal magnesium at 0.8 mmol/L (0.6–1.2 mmol/L), low 25[OH]D at 24 nmol/L (25–200 nmol/L) and PTH at 0.9 pmol/L (1.1–5.6 pmol/L), and inappropriately normal spot urinary calcium/creatinine ratio at 0.36 (0.06–0.45). Family history was significant for a maternal aunt having isolated hypoparathyroidism. The patient was initially commenced on calcium carbonate and calcitriol supplementation with a thiazide diuretic added later due to worsening hypercalciuria (calcium/creatinine ratio 0.98). CaSR gene analysis detected a novel missense mutation in exon 7, c.2431A > G (M811V) (Figure 1), which was subsequently shown to segregate with the hypocalcaemic phenotype in other family members on screening (Figure 2). 5
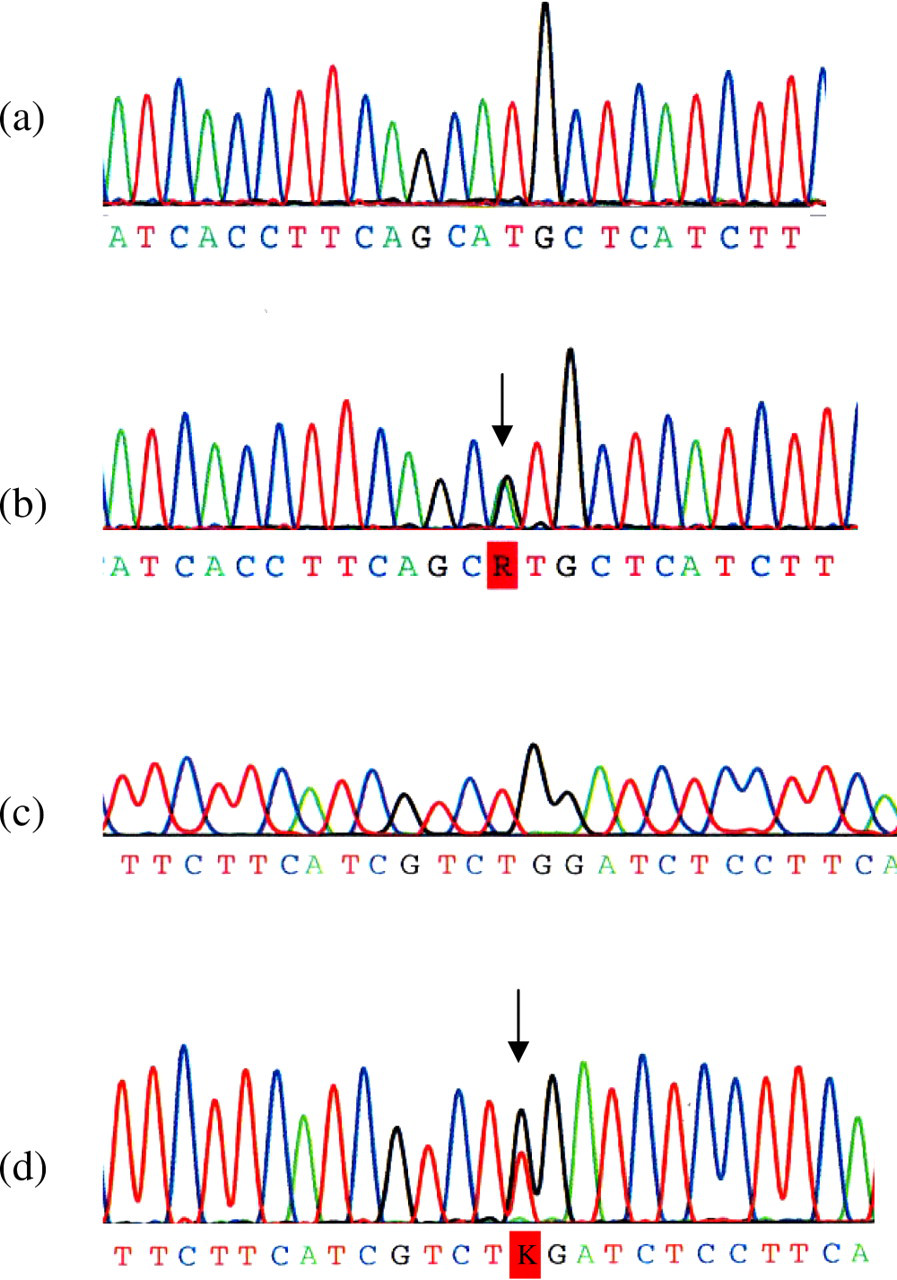
(a) Wild-type sequence, compared with mutated sequence in panel b for case 2, having an NM_000388.2: c.2431A > G missense mutation. This results in a change in sequence from ATG to GTG, resulting in the substitution of a methionine residue with a valine residue (p.Met811Val). Panel c shows wild-type sequence, compared with mutated sequence in panel d for case 4, having an NM_000388.2: c.2453G > T missense mutation. This results in a change in sequence from TGG to TTG resulting in the substitution of a tryptophan with a leucine residue (p.Trp818Leu)
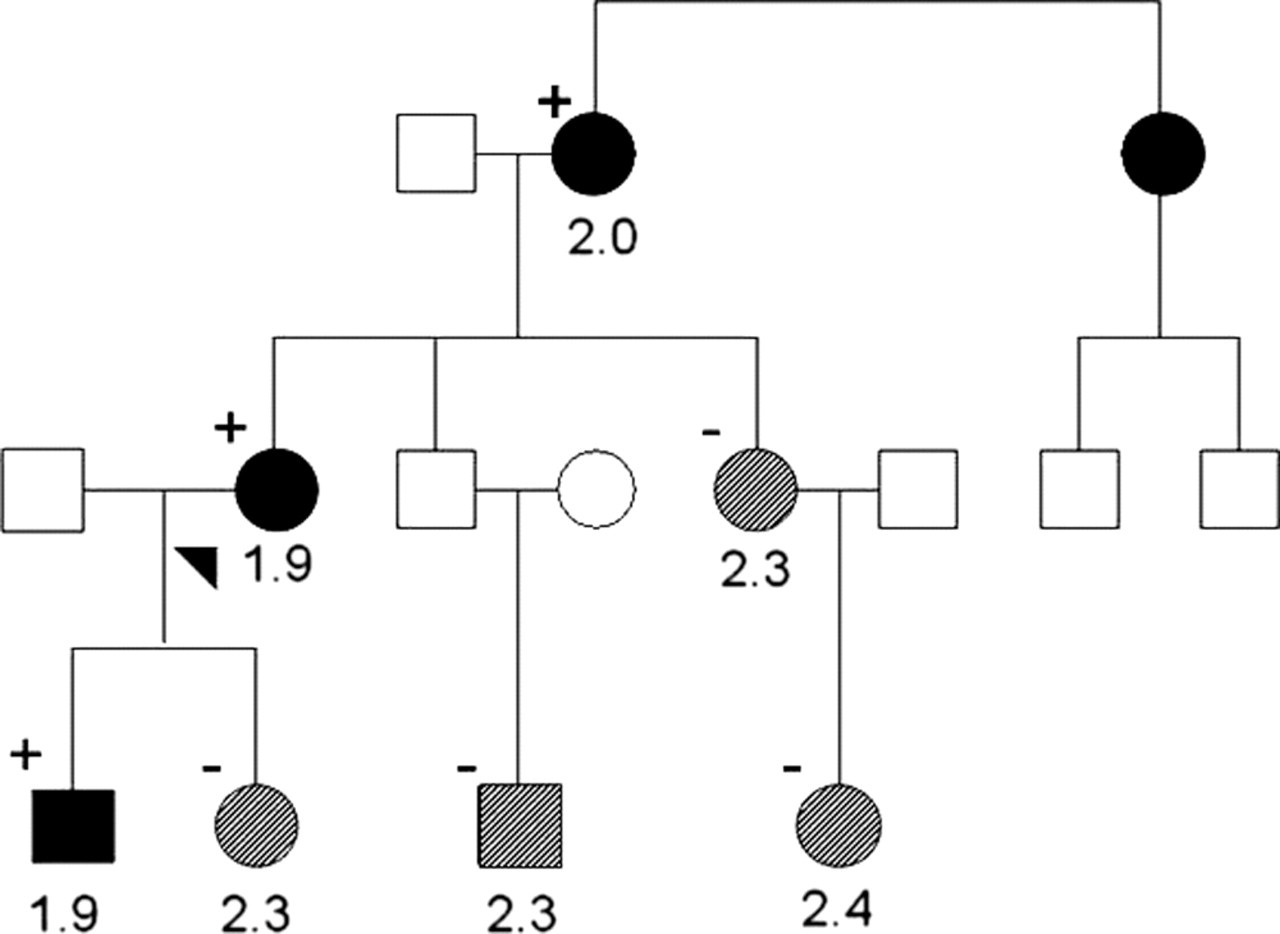
Family pedigree chart for case 2. Individuals are represented by squares (male) and circles (female) with the proband marked with an arrowhead. Clinical status is indicated by filled symbols (hypocalcaemic), shaded symbols (normocalcaemic) and open symbols (unknown). Serum calcium values (normal range 2.2–2.6 mmol/L) and the presence (+) or absence (−) of the c.2431A > G mutation in tested family members is shown. DNA from the affected aunt was unavailable for genetic testing
Case three
A 27-year-old woman was admitted to hospital following a collapse with brief loss of consciousness four hours after taking her first dose of citalopram. She was hypotensive with blood pressure (BP) 90/70 and electrocardiogram (ECG) demonstrated borderline prolongation of QTc at 452 ms. Electrolytes demonstrated low corrected calcium of 1.8 mmol/L (2.2–2.6 mmol/L), with normal phosphate of 1.2 mmol/L (0.8–1.4 mmol/L), magnesium of 0.9 mmol/L (0.6–1.2 mmol/L) and creatinine of 0.06 mmol/L (0.05–0.11 mmol/L). PTH was inappropriately normal at 3.8 pmol/L (1.6–7.0 pmol/L), 25[OH]D was 50 nmol/L (50–150 nmol/L) and urinary calcium excretion inappropriately normal at 5.8 mmol/24 h (2.5–7.5 mmol) at a time when plasma calcium was 1.9 mmol/L. There was no history of neck surgery or family history of hypocalcaemia. Her citalopram was ceased with normalization of her QTc and she was commenced on calcium carbonate and calcitriol supplements. CaSR mutational analysis confirmed her to be heterozygous for a previously described c.372C > A (N124 K) mutation. 6 Asymptomatic hypocalcaemia with relative hypercalciuria was identified in her mother and sister on family screening, neither of whom had required treatment.
Case four
A 20-year-old woman was referred for investigation of hypocalcaemia. She gave a history of carpo-pedal spasm and hypocalcaemic neonatal seizures. There was no ectopic calcification, vitiligo, alopecia or evidence of chronic fungal infections. There was carpal spasm after 75 s of forearm ischaemia and the Chvostek sign was positive. ECG showed a prolonged QT interval. Corrected calcium concentrations of 1.8, 1.69 and 1.67 mmol/L (2.2–2.6 mmol/L) had been documented over the last year with phosphates of 1.8, 1.4 and 1.7 mmol/L (0.8–1.4 mmol/L). Serum creatinine was normal and spot urine calcium/creatinine ratio elevated at 1.95 (0.06–0.45). Serum magnesium was 0.66 mmol/L (0.75–1.0 mmol/L). With a low-normal PTH concentration of 1.1 pmol/L (1–5 pmol/L), the patient was assigned a diagnosis of idiopathic hypoparathyroidism and commenced on calcium supplements and calcitriol 0.25 μg daily. Subsequently she developed mild renal impairment (estimated glomerular filtration rate 60 mL/min) and nephrocalcinosis and was switched to calciferol 1.25 mg monthly together with the PTH analogue teriparatide (20 μg twice a day) with normalization of serum calcium. Following identification of hypocalcaemia in her daughter, she underwent mutation analysis of the CaSR gene, which showed that she was heterozygous for the novel mutation c.2453G > T (W818L) (Figure 1).
Methods
All coding exons and flanking intronic regions of the CaSR gene were amplified by polymerase chain reaction (PCR) and were analysed by automated fluorescent sequencing. DNA sequence was compared with the Genbank reference sequence (GenBank NC_000003.10; range 123385220–123488032). Sequence nomenclature is based on the mRNA coding reference sequence for the CaSR gene (GenBank NM_000388.2). The following primer pairs were used for PCR amplification and DNA sequencing: Exon 2F and 2R: 5′ TGCCCTGGAGAGACGGCAGA 3′, 5′ AGAGATTGGCAGATTAGGCC 3′ Exon 3F and 3R: 5′ CCCATTTTCTTCCACTTCTT 3′, 5′ CCCGTCTGAGAAGGCTTGAGT 3′ Exon 4F and 4R: 5′ CATGTTCTTGGTTCTCTCCA 3′, 5′ GCTATATAAGTGAAGGACGC 3′; Exon 5F and 5R: 5′ GTACTCATTCTTTGCTCCTC 3′, 5′ CTGGTTTTCTGATGGACAGC 3′ Exon 6F and 6R: 5′ CAAGGACCTCTGGACCTCCCTTTGC 3′, 5′ GACCAAGCCCTGCACAGTGCCCAAG 3′ Exon 7F and 7R: 5′ CATGCTAGACCTCTGGTGTGCAGG 3′, 5′ TCTTCCTCAGAGGAAAGGAGTCTGG 3′.
The following additional primers were used for sequencing exons 4 and 7: Exon 4: forward: 5′ GCCAAAGTCATCGTGGTTTT 3′, 5′ GCCAAGGAGTTTTGGGAAGA 3′; reverse: 5′ CCAGATCTTGCCCGTGATAT 3′ Exon 7: forward: 5′ ACCAACCGAGAGCTCTCCTA 3′, 5′ TGCCTGCTGGCTGCCATCTG 3′, 5′ GATGCAAGCAGAAGGTCATC 3′; reverse: 5′ GCAGAGCAGGGAGAAGAGGA 3′, 5′ CCGGGACTTGAAGGCAAAGA 3′, 5′ CAGTGAGAAGGTGACCGTGC 3′.
Discussion
The CaSR is a member of the seven transmembrane, G protein-coupled receptor superfamily. Initially identified in parathyroid tissue, receptor tissue distribution is now known to include calcitonin-secreting C-cells of the thyroid, renal tubules, osteoblasts and enterocytes, other tissues important in the maintenance of calcium homeostasis. 7–9 CaSR mutations, while rare, have the potential to cause hyper- or hypocalcaemia, manifesting as familial hypocalciuric hypercalcaemia, neonatal severe hyperparathyroidism or ADHH. The CaSR single-nucleotide polymorphism R990G is associated with primary hypercalciuria in individuals with and without a history of renal calculi. 10,11
Since mutational analysis of the CaSR gene became available, many individuals with hypocalcaemia have had their diagnosis revised. Lienhardt et al. 12 reported a 42% prevalence of activating CaSR mutations in unrelated individuals with isolated hypoparathyroidism and Gunn and Gaffney 13 estimated the population prevalence of this condition to be one in 70,000.
Biochemical features which may suggest a diagnosis of ADHH include family history of hypocalcaemia, PTH within or just below the normal range and hypomagnesaemia. In the absence of a family history the possibility of de novo mutations needs to be considered although it is more likely that family members are affected but remain unrecognized. While ADHH is characterized by inappropriate hypercalciuria, there is no reliable cut-off for urinary calcium below which the diagnosis is excluded. 12,13 Clinical presentation is variable and although commonly asymptomatic, patients may present with neuromuscular irritability, seizures, laryngospasm, renal dysfunction or calculi, left ventricular failure or basal ganglia calcification. 12,14
The kidney plays an important role in regulating calcium homeostasis and under normal physiological conditions <1% of the filtered load is excreted in urine. 15 Calcium absorption takes place throughout the nephron with 80–85% occurring passively secondary to paracellular movement down an electrochemical gradient. PTH, calcitonin and 1,25[OH]2D stimulate transcellular absorption in the distal convoluted tubule (DCT) and thick ascending limb (TAL) of the loop of Henle. Calcium crosses the apical membrane through calcium-selective channels (TRPV5) with subsequent energy-dependent efflux across the basolateral membrane via Ca2+ – ATPase (PMCA) and Na+/Ca2+ exchangers.
Activation of the renal CaSR modifies calcium, magnesium and phosphate reabsorption. Receptor stimulation inhibits the renal medullary potassium channel (Kir1.1), 16 primarily located in the apical membrane of the TAL, and the inwardly rectifying basolateral potassium channels of the DCT (Kir4.1 and Kir4.2). 17 The fall in tubular potassium limits NaCl reabsorption via the Na+-K+-2Cl− transporter. The decrease in transepithelial electrical potential, usually maintained by recycling of potassium into the tubule and movement of absorbed chloride across the basolateral cell membrane, reduces passive paracellular cation movement resulting in increased urinary calcium and magnesium excretion. In addition, PTH-sensitive calcium reabsorption in the cortical TAL is inhibited by CaSR activation by an unknown mechanism.
Hyperphosphataemia is commonly seen in ADHH and may occur despite PTH concentrations in the normal range. 18 Activation of the CaSR in the proximal renal tubule inhibits PTH-induced internalization of the sodium–phosphate co-transporter, thereby promoting phosphate reabsorption. 19 Reduced water permeability of the inner medullary collecting duct is an additional consequence of CaSR stimulation, secondary to inhibition of vasopressin-induced incorporation of aquaporin-2 water channels into the apical membrane.
At least 85 different activating mutations of the CaSR have been characterized to date (
Vitamin D response elements have been identified in the promoter sequence of the CaSR with 1,25[OH]2D shown to upregulate gene expression in the parathyroid, thyroid C-cell and renal tubules. 20 1,25[OH]2D supplementation would be expected to increase urinary calcium excretion and in the setting of ADHH this treatment may fail to adequately raise serum calcium while potentially contributing to nephrocalcinosis, nephrolithiasis and renal damage (as illustrated by case four). 18 Treatment of ADHH with calcium and vitamin D supplements should therefore be reserved for symptomatic individuals, the aim being to raise serum calcium to the level at which symptoms abate. Vitamin D dose should be kept as low as possible. Life-long follow-up is necessary with close monitoring of serum and urine calcium. Nephrocalcinosis may develop despite maintenance of serum calcium below the lower limit of the reference range and in patients with frank hypercalciuria, or relative hypercalciuria complicated by renal calculi, the addition of a thiazide diuretic, synthetic PTH, or in the future a CaSR antagonist (calcilytic), may be beneficial. 12,21,22
We have described four cases of ADHH. While the association between hypocalcaemia and the C129Y and N124K CaSR mutations has previously been reported,
4,6
the M811V and W818L mutations are novel. No other mutations or polymorphisms of the CaSR gene were identified in any of the four cases. The M811V and W818L mutations occur within the transmembrane domain of the receptor protein, resulting from the substitution of methionine with valine and tryptophan with leucine, respectively. Methionine and valine are both hydrophobic residues and therefore this base substitution would not be expected to alter receptor conformation. However, biochemical and genetic screening of family members has confirmed co-segregation of the M811V mutation with hypocalcaemia. The tryptophan residue at position 818 is highly conserved across species with Polyphen (
In summary, the CaSR plays an important role in maintaining serum calcium concentrations within a narrow physiologic range. Distinguishing isolated hypoparathyroidism from ADHH is clinically important due to the risk of renal calcium deposition and subsequent renal dysfunction in the latter. While a family history of hypocalcaemia with autosomal dominant inheritance or the presence of certain biochemical abnormalities may provide diagnostic clues, differentiation of ADHH from other causes of isolated hypoparathyroidism remains challenging and gene sequencing should be considered. After identification of an index case, measurement of serum calcium in family members is a sensitive and inexpensive screening tool.
DECLARATIONS