
Abstract
Autosomal dominant hypercholesterolaemia is genetically heterogeneous, but most commonly (∼93%) caused by mutations in low-density lipoprotein receptor (LDLR), where the disease is known as familial hypercholesterolaemia (FH), or apolipoprotein B-100 (APOB) (∼5.5%), where the disease is known as familial defective APOB (FDB), while in ∼2% of patients the mutation is in the proprotein convertase subtilisin/kexin type 9 gene. Homozygous FH having inheritance of two LDLR mutations is a rare but recognized syndrome associated with an extreme hypercholesterolaemia and early-onset coronary artery disease. We present a 15-year-old girl with untreated total cholesterol levels of 8.8 mmol/L who was heterozygous for both the LDLR p.Leu479Pro and APOB p.Arg3527Gln mutation. Cascade testing confirmed the paternal origin of the LDLR mutation and revealed a maternal diagnosis of FDB. This case provides further evidence that the combined effect of an LDLR and an APOB mutation give rise to a phenotype more severe than either mutation alone and is more severe than homozygous FDB, but less severe than homozygous FH. It also highlights the need to consider the presence of additional mutations in families where relatives have varying phenotypes.
Introduction
The term familial hypercholesterolaemia (FH) is commonly used to refer to any dominant autosomal gene disorder characterized by increased plasma levels of total cholesterol (TC) and low-density lipoprotein cholesterol (LDL-C), tendon xanthoma and premature symptoms of coronary heart disease. 1 However, given that the disorder is known to be caused by mutations in at least three different genes, this has the potential to cause some confusion, particularly in the context of genetic analysis where clarity is required for confirmatory diagnosis and appropriate cascade testing of relatives. A preferred general term is autosomal dominant hypercholesterolaemia, with FH being reserved for the most common form of the disease due to mutations in the LDL receptor (LDLR) gene that accounts for ∼93% cases in the UK. Mutations in the apolipoprotein B-100 gene (APOB) account for ∼5% cases, in which case the disease should be referred to as familial defective APOB (FDB). Other more rare genetic causes may be referred to by their associated gene, for example mutations in the proprotein convertase subtilisin/kexin type 9 gene (PCSK9) gene that accounts for ∼2% cases in the UK may be referred to as PCSK9-related ADH. 2,3
There are no specific clinical criteria to differentiate between these forms, with individuals heterozygous for either a mutation in the LDLR or APOB gene being shown to have similar presentations, with TC levels in FDB subjects being in the range 7.5–9.0 mmol/L, 4 while patients with mutations in PCSK9 tend to have more severe disease. 5 Around one in a million live births are homozygous for LDLR mutations and typically present with very severe hypercholesterolaemia (i.e. TC levels 15–20 mmol/L), and early-onset disease. 1 Homozygous FDB is predicted to occur at around one in four million individuals, who have plasma TC levels in the range 10–16 mmol/L, which is therefore less severe than those with homozygous FH and is more comparable to FH heterozygotes (where TC is in the range 7.5–10 mmol/L). To date, 10 individuals from five families have been reported who are heterozygous for both an LDLR and APOB mutation. 6–9
We present the case of a 15-year-old girl who was referred because of a family history of premature coronary artery disease and hypercholesterolaemia.
Materials and methods
Family details
The girl was referred to a lipid clinic at age 13 years because of TC of 8.8 mmol/L when she was tested at age 12 years following the death of her father at age 48 years from a sudden myocardial infarction. She had never smoked, was physically active and kept to a good diet. On examination she had no tendon xanthoma or other clinical features of hyperlipidaemia. Her father was reported to have had a TC of 10.6 mmol/L and commenced on statin treatment at age 35 years. Her paternal grandmother was found to found to have a TC of 13 mmol/L at the age of 60 years through a ‘well person’ screen and was subsequently commenced on statins. She is now 78 years old and neither she nor her son had tendon xanthoma. The girl was advised to follow a low fat diet, take regular exercise and was commenced on atorvastatin. The lipid profile and mutation analysis of samples from the girl and her relatives are shown in Table 1.
Lipid profile and DNA test results
*Results obtained when undergoing treatment with atorvastatin 20mg
p.Leu479Pro heterozygote is in LDLR
p.Arg3527Gln heterozygote is in APOB
Molecular analysis
DNA extraction, ARMs analysis (testing for 20 common FH-causing mutations in the UK, including 18 LDLR mutations, APOB p.Arg3527Gln and PCSK9 p.Asp374Tyr) and sequence analysis were performed as described previously. 10–12 Restriction analysis to confirm the presence of the p.Arg3527Gln mutation was carried out as described by Mamotte et al. 13
Results
DNA analysis indicated the girl to be heterozygous for both the p.Leu479Pro in the LDLR gene and the p.Arg3527Gln APOB mutation. Subsequent analysis of her siblings and their mother showed that her elder brother and mother are heterozygous for APOB p.Arg3527Gln and none have the LDLR p.Leu479Pro mutation. This confirmed the maternal origin of the APOB mutation, and led to the diagnosis of FDB in the mother and her eldest son, but reduced the likelihood of ADH in the youngest son and daughter (see Figure 1, pedigree).The eldest son had previously had his cholesterol levels tested and was presumed to be unaffected as he had a TC of 5.6 mmol/L. On repeat cholesterol testing his LDL-C levels were found to be just above the age/sex-specific cut-off for members of a family in whom FH had been positively identified. His latest TC is 5 mmol/L; he has an extremely healthy diet and lifestyle and is not currently taking statins. Samples from the proband's two paternal male cousins were subsequently tested for the LDLR mutation and one was found to be heterozygous for this mutation, thus providing evidence for the paternal origin of the p.Leu479Pro LDLR mutation.
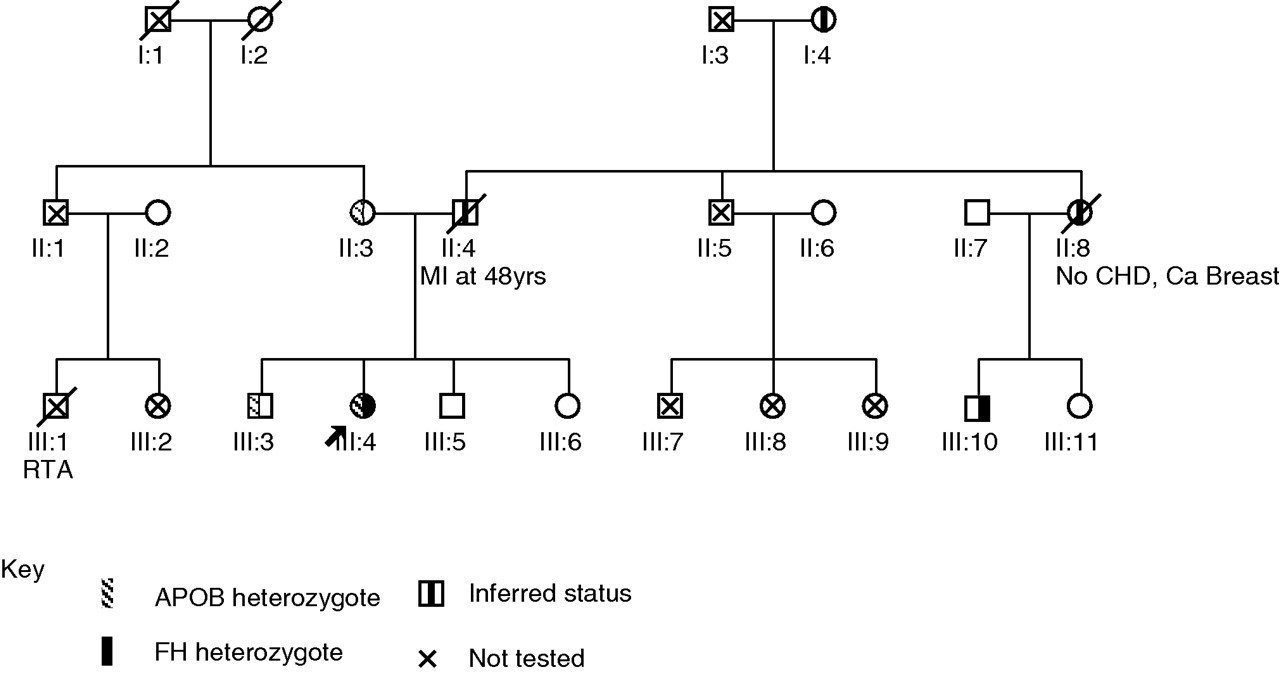
Family pedigree
Discussion
Given that the frequency of FH and FDB in the European population is around 1/500 and 1/1000, respectively, 1–4 double heterozygotes, as in this young woman, would be expected to be present at around 1/1,400,000 (around 40 cases in the UK population). However, only five families have been reported in the literature to date. 6–9 Rubinsztein et al. 7 reported a South African family who had five individuals who were double heterozygotes for the p.Asp227Glu LDLR and p.Arg3527Gln APOB mutations and one individual who was a ‘complex’ heterozygote for both these mutations plus the p.Val429Met LDLR mutation. The individuals who were double heterozygotes were reported to have lipid levels and clinical features that were intermediate in severity between heterozygous and homozygous FH. Shyong Tai et al. 8 reported a family in which an individual was heterozygous for the p.Asn428Lys LDLR mutation and p.Arg3527Trp APOB mutation. The proband was found to have a pretreatment serum LDL-C level approximately twice as high as her siblings. Benlian et al. 9 reported two unrelated French patients who were heterozygous for the p.Arg3527Gln APOB mutation and either the Trp66Gly (HGVS p.Trp87Gly) or Glu207Lys (HGVS p.Glu228Lys) LDLR mutations. The probands were reported to have an unusual phenotype of aggravated hypercholesterolaemia that was complicated with premature coronary arterial disease, although remaining responsive to lipid-lowering drugs. It was stated that this phenotype was distinct from that observed in the proband's heterozygous relatives and also from those seen in both FH and FDB homozygotes.
The case we present in this paper is a double heterozygote for the p.Leu479Pro mutation in the LDLR gene and the common p.Arg3527Gln mutation in the APOB gene. The p.Leu479Pro mutation has been reported previously, 5,14,15 and we have found this mutation in 4/414 (∼1%) of our unrelated patients with detected mutations. Although no functional studies have been performed to confirm its pathogenicity, the p.Leu479Pro mutation occurs in the epidermal growth factor spacer domain of the LDLR protein that is required for acid-dependent dissociation of the receptor from its ligand during recycling, and the mutation may interfere with receptor recycling. The leucine at codon 479 is highly conserved between species, being a leucine in all sequenced mammalian LDLR genes, and bioinformatic tools such as PolyPhen and SIFT predict that the mutation will have a detrimental effect on receptor function.
The APOB p.Arg3527Gln mutation accounts for the vast majority 5 of cases of FDB and has been shown to reduce binding to around 30% of LDL in vitro. 16,17 Although the mutation is not located within the LDL-receptor binding site (3386–3396), p.Arg3527Gln appears to cause improper protein folding and reduced receptor binding.
The presence of two mutations was not suspected clinically in our patient and was identified only as a result of the genetic screening. In view of her good response to statins, it could have remained undetected.
Despite the exaggerated hypercholesterolaemia, individuals who are double heterozygotes for both FDB and FH have been reported to retain the ability to respond to cholesterol-lowering agents such as statins. This is also seen in our patient, as treatment with atorvastatin 10 mg daily reduced her TC to 6.6 mmol/L and an increase to 20 mg resulted in a TC level of 5.8 mmol/L.
The existence of both an LDLR and APOB mutation should be considered in families (a) where an LDLR mutation is present in some but not all individuals who exhibit hypercholesterolaemia and (b) in individuals who have the same LDLR mutation but have an exaggerated phenotype compared with other members of the family. Thus, in cascade DNA testing of relatives it may be useful to include other common mutations besides the known family mutation. This does, however, require that the family be appropriately counselled, but has similarities to strategies used for genetic screening for cystic fibrosis and familial breast cancer. In addition, it is possible that individuals may also be a double heterozygote for other ADH mutations. Two cases of LDLR and PCSK9 double heterozygotes have been reported by Piscciata et al. 18 The LDL-C level in the double heterozygotes from these families was 56% and 44% higher than those relatives who were just heterozygous for the LDLR mutation. Thus, missense mutations in PCSK9 may also worsen the clinical phenotype of patients carrying an LDLR mutation.
It therefore appears that there is a new class of patients with digenic lipid disorders who have clinical features that result from the combined effects of two independent loci.
DECLARATIONS