
Abstract
Statins are agents widely used to lower LDL-cholesterol (LDL-C) in primary and secondary prevention of coronary heart disease. The five statins available in the UK (simvastatin, pravastatin, fluvastatin, atorvastatin and rosuvastatin) differ in many of their pharmacologic properties. In addition to lowering LDL-C, statins also increase HDL-cholesterol (HDL-C) moderately. There have been rare reports of significant HDL-C decreases in patients commenced on fibrates and when thiazolidinediones are added to fibrates. This is known as a ‘paradoxical HDL-C decrease’ as both groups of agents usually increase HDL-C. This phenomenon has never been clearly documented following statin therapy. We now describe a patient with type 2 diabetes who showed this paradoxical fall in HDL-C (baseline HDL-C: 1.8 mmol/L; on simvastatin 40 mg HDL-C 0.6 mmol/L; on atorvastatin 20 mg HDL-C 0.9 mmol/L) with a similar decrease in apolipoprotein A1. No similar decrease was observed with pravastatin and rosuvastatin therapy. This phenomenon appeared to be associated with statin treatment and not a statin/fibrate combination. Our patient clearly demonstrated a paradoxical HDL-C fall with simvastatin and atorvastatin, but not pravastatin or rosuvastatin. Simvastatin and atorvastatin share many pharmacokinetic properties such as lipophilicity while pravastatin and rosuvastatin are relatively hydrophilic and are not metabolized by cytochrome P450 3A4. However, these characteristics do not explain the dramatic reductions in HDL-C observed.
Introduction
Since the 4S trial 1 statins have been an integral part in the prevention of cardiovascular disease. They inhibit 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMG CoA reductase) leading to increased expression of hepatic LDL receptors increasing LDL uptake. 2 The commonly used statins vary in pharmacokinetics, efficacy and tolerability. 3,4
Simvastatin, atorvastatin and fluvastatin are lipophilic while pravastatin and rosuvastatin are hydrophilic. 5,6 Some statins are metabolized by the CYP P450 enzymes (simvastatin and atorvastatin by cytochrome P450 3A4 [CYP3A4] and fluvastatin by cytochrome P450 2C9 [CYP2C9]) in the liver to active metabolites. 7 Pravastatin and rosuvastatin are not substantially metabolized this way. Simvastatin, atorvastatin and fluvastatin are excreted in bile while pravastatin and rosuvastatin are eliminated unaltered via biliary and renal routes. 8,9
Statins can decrease triglycerides (TG) in some patients by reducing VLDL synthesis and increasing clearance.
10,11
They have been shown to lower TG concentrations between 10% and 20%; greater decreases are seen in patients with higher baseline concentrations.
12
Statins have been shown to increase apolipoprotein A1 (ApoA1) and HDL-cholesterol (HDL-C) concentrations.
13,14
However, there does not appear to be an association between LDL-cholesterol (LDL-C) lowering and increases seen in HDL-C.
12
Meta-analysis of the VOYAGER database, which included 37 studies and 32,258 individuals on simvastatin, atorvastain and rosuvastatin, showed that HDL-C increases with simvastatin and rosuvastatin were comparable and superior to those seen with atorvastatin. Interestingly, the HDL-C increase positively correlated with increasing doses of simvastatin and rosuvastatin, but not with increasing doses of atorvastatin.
12
Further, percentage changes in ApoA1 correlated with that of HDL-C. It was also observed that diabetes is associated with significantly smaller increases in HDL-C on statin treatment (with the exception of atorvastatin 80 mg). This observation has previously been seen with simvastatin treatment in the HPS study.
15
We describe here a case where significant decreases in HDL-C and ApoA1 were seen in a patient with type 2 diabetes with simvastatin and atorvastatin, but not pravastatin or rosuvastatin.
Methods
Lipids were measured on the Roche Modular P800 analyser using Roche reagents by enzymatic colorimetric methods (total cholesterol [TC] and HDL-C by direct qualitative methods). ApoA1 was analysed on the Vitros 5.1FS chemistry system using Vitros chemistry products. The addition of antisera specific for human ApoA1 yielded antigen–antibody complexes and ApoA1 concentrations were measured via turbidimetry. High-performance liquid chromatography was used to estimate the HbA1c using the TOSOH HLC-723G7 automated glycohemoglobin analyser. No changes were made to any of the assays during our patient's follow-up period.
Case history
A 56-year-old woman with type 2 diabetes mellitus diagnosed eight years previously was referred to the metabolic clinic with a HDL-C of 0.6 mmol/L. She was on metformin 2 g and simvastatin 40 mg and experienced myalgia. At the time of referral HbA1c was 6.8%, TC 4.8 mmol/L and TG 1.5 mmol/L (Table 1, row b). No pretreatment lipid results were available at the time and the patient was regarded as having the metabolic syndrome with her healthy lifestyle controlling the TG concentrations. Her alcohol intake was around 10 units weekly and did not vary significantly. In view of the low HDL-C and myalgia (creatinine kinase was not measured at that visit), simvastatin was discontinued and fenofibrate 160 mg prescribed. It was six months prior to the publication of the FIELD study. 16 Our patient's HDL-C was much lower than the mean baseline HDL-C in the FIELD study (1.1 mmol/L); we may have started fenofibrate anyway. Ten weeks later, the HDL-C had risen to 1.2 mmol/L (TC 6.3 mmol/L, TG 0.9 mmol/L – Table 1, row c).
Details of lipid-lowering medication and lipids, ApoA1, ApoB and HbA1c concentrations
ApoA1, apolipoprotein A1; TC, total cholesterol; TG, triglycerides; HDL-C, HDL-cholesterol; LDL-C, LDL-cholesterol
All lipid measurments were carried out on fasted samples
*LDL-C was calculated not measured
As the TC and calculated LDL-C did not reach the Joint British Societies 2 optimal targets (TC < 4 mmol/L and/or LDL-C < 2 mmol/L) 17 atorvastatin 10 mg was added to the fenofibrate. It was noted that the HDL-C had dropped to 0.5 mmol/L (TC 4.7 mmol/L, TG 1.1 mmol/L – Table 1, row d). The results were re-checked and the HDL-C and ApoA1 were 0.9 mmol/L and 90 mg/dL, respectively (Table 1, row e). We considered the possibility of paradoxical HDL-C decrease due to statins, which we had experienced previously with fibrates and thiazolidinediones. The atorvastatin was then discontinued.
We obtained the pretreatment results from the pathology archives (TC 5.7 mmol/L, TG 1.0 mmol/L, HDL-C 1.8 mmol/L – Table 1, row a), which suggested that the HDL-C reduction was associated with statin treatment. Six weeks after atorvastatin was discontinued, the HDL-C and ApoA1 had increased to 2.0 mmol/L and 142 mg/dL, respectively (Table 1, row g). As the TC was higher than the target at 5.4 mmol/L, we started pravastatin 10 mg and the fenofibrate was discontinued. Two months later, the HDL-C was 2.3 mmol/L, i.e. pravastatin did not induce a decrease (TC 4.8 mmol/L, TG 0.7 mmol/L, ApoA1 153 mg/dL – Table 1, row h). The pravastatin was increased to 20 mg and the HDL-C and ApoA1 remained unaltered. Since the TC and LDL-C were still above target, pravastatin was stopped and rosuvastatin 5 mg was introduced and subsequently increased to 10 and 20 mg. No significant reduction in HDL-C or ApoA1 was noted (Table 1, rows l–p).
A chronology of the above results is presented in Figure 1. During this period the HbA1c ranged between 6.3 and 7.2%.
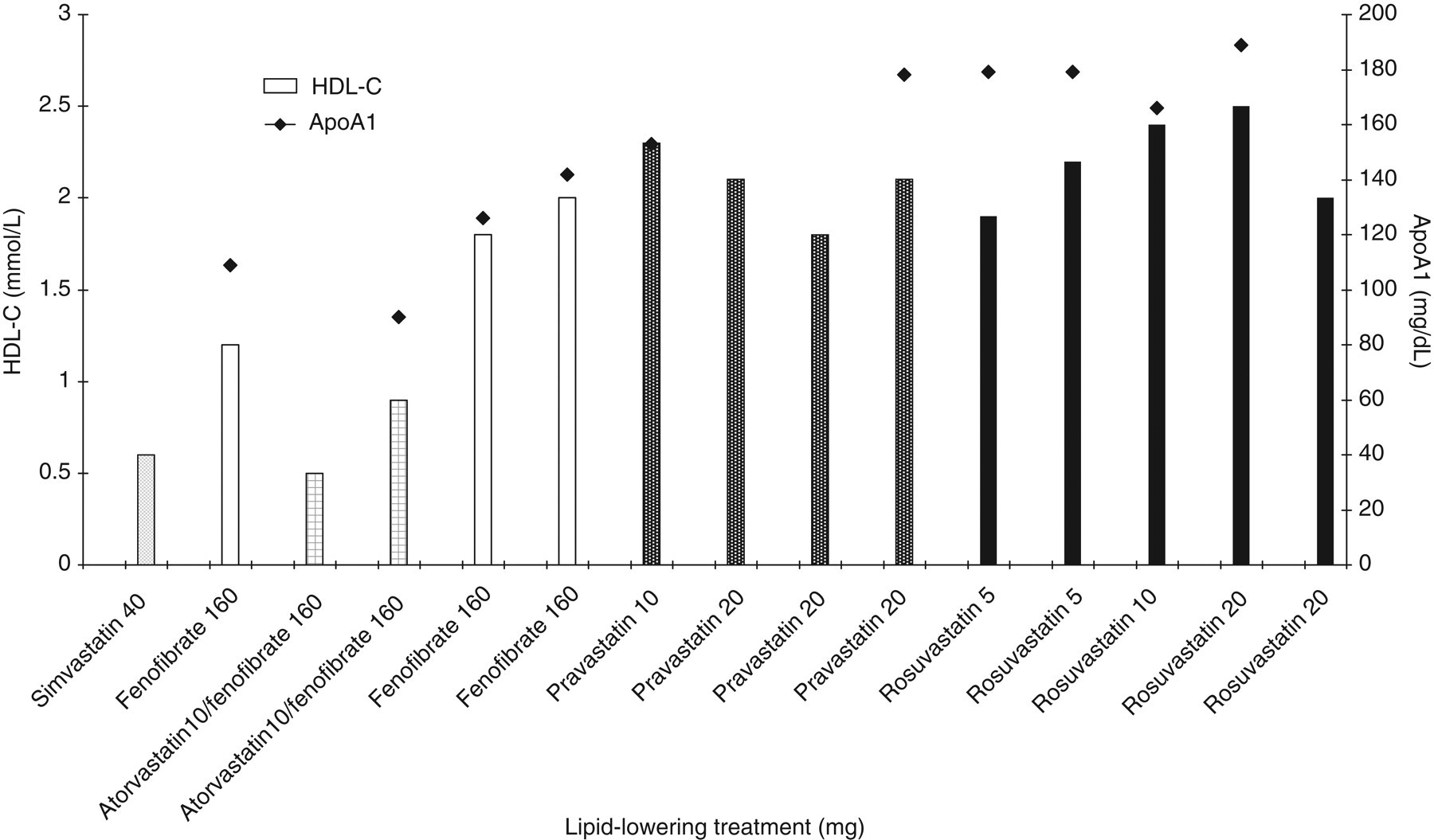
The HDL-C, ApoA1 results and treatment details including the different statins and doses are shown in this figure. ApoA1, apolipoprotein A1; HDL-C, HDL-cholesterol
Our patient had two brothers who were on atorvastatin. Each had their lipid profile checked and HDL-C concentrations were 1.9 and 1.2 mmol/L, respectively.
Discussion
Our patient demonstrated a marked reduction in HDL-C and ApoA1 to two of the four statins tried. Simvastatin and atorvastatin that resulted in the decrease in both ApoA1 and HDL-C have many features not shared by pravastatin and rosuvastatin. Pravastatin and rosuvastatin are hydrophilic compared with simvastatin and atorvastatin. 5,6 Pravastatin is not metabolized by the CYP450 enzymes and rosuvastatin shows limited metabolism with nominal active metabolites 9 while atorvastatin and simvastatin are metabolized by CYP450 3A4 leading to active metabolites. 7
Paradoxical HDL-C and ApoA1 decreases have been reported with fibrates and both fibrate/statin and fibrate/thiazolidinedione treatment. 18–20 When our patient initially presented with a HDL-C concentration of 0.6 mmol/L she was not on a fibrate. This suggests that simvastatin and atorvastatin either directly or via their metabolites had an effect on ApoA1 and HDL-C metabolism.
Low concentrations of HDL-C and ApoA1 are often observed in patients with either overproduction or impaired clearance of TG-rich lipoproteins; 21 our patient however did not demonstrate hypertriglyceridaemia. ApoA1 is synthesized in the liver and intestine, and unless free cholesterol and phospholipid uptake occurs rapidly it undergoes rapid catabolism. 22 Thus, genetic mutations reducing the function of ApoA1, ATP binding cassette transporter 1 (ABCA1), phospholipid transfer protein (PLTP), lecithin-cholesterol acyltransferase (LCAT), peroxisome proliferator-activated receptors alpha (PPAR-α) genes or increasing functionality of cholesteryl ester transfer protein (CETP), scavenger receptor class B1 (SRB1) or hepatic lipase would lead to low concentrations of ApoA1 and HDL-C. As low HDL-C and ApoA1 coincided only with two of the statins, it is unlikely that no underlying functional genetic mutation exists in our patient. However, functionality at one or more of the above points of control could have been altered. Simvastatin possesses a very short half-life (approximately 2 h) and even four months after it was discontinued the HDL-C had only risen to 1.2 mmol/L. This was lower than the pretreatment concentrations (1.8 mmol/L) and that seen later. However, after discontinuing atorvastatin the increase in HDL-C was more rapid reaching 1.8 mmol/L after three weeks. It is possible that both simvastatin and atorvastatin might have affected gene expression/transcription of proteins involved in HDL-C metabolism. However, the relatively rapid increase of HDL-C following atorvastatin withdrawal is not consistent with this.
Our patient's glycaemic control remained stable during the period where the changes in HDL-C were observed. Her diabetes treatment was not altered and thus it is unlikely that the HDL-C change was due to changes in insulin resistance.
With the available data we are unable to ascertain the mechanism of the paradoxical HDL-C reduction observed. It is clear that statins are likely to have a role beyond LDL-C reduction and functional differences between the available drugs are possible, at least in certain patients. This case highlights the importance of measuring HDL-C before and after the introduction of a statin or after a change to another statin.
DECLARATIONS