
Abstract
Background
Circulating oxidized low-density lipoproteins (LDLs) (ox-LDLs) could be a sensitive marker to predict future cardiovascular events. However, a method to evaluate oxidized forms of LDLs systemically in human plasma is not yet established. In this study, we developed a novel and convenient high-performance liquid chromatography (HPLC) method for measuring ox-LDL levels in humans.
Methods
Human plasma lipoproteins were separated by a modified HPLC method using a diethylaminoethyl-type anion-exchange gel column with stepwise elution. Ox-LDLs were detected by postcolumn reaction with a regent containing cholesterol esterase and cholesterol oxidase. Particle size of each LDL fraction separated by HPLC was determined in 61 healthy subjects.
Results
Our HPLC method separated LDLs into three fractions, which were designated as LDL-1, LDL-2 and LDL-3, on the basis of their negative charges, with LDL-3 the most strongly retained fraction migrating fastest in the anodic direction, a property that reflects the net negative charge of the molecule. Western blot analysis revealed that apolipoprotein B100 in LDL-3 fraction was the most fragmented and oxidatively modified. When LDLs were oxidized in vitro by Cu2+ or 2,2-azo-bis (2-aminopropane)-2HCl or modified by various aldehydes, all of the LDL fractions migrated at the position of LDL-3. Further, among three fractions, particle size was smallest in LDL-3 fraction.
Conclusion
Here, we developed a convenient HPLC method and identified LDL-3 as oxidized LDL fractions, although ox-LDLs were present in LDL-2 fraction, albeit lesser concentrations than in LDL-3 subfraction. Measuring ox-LDL levels in human plasma by this method may be useful to evaluate atherosclerotic disorders.
Introduction
There is a growing body of evidence that modified LDLs, especially oxidized LDLs (ox-LDLs), could play an important role in the development and progression of atherosclerosis. 1–3 Ox-LDLs are present in human serum or plasma, and the levels are increased in patients with atherosclerotic disorders, such as coronary heart disease, stroke and peripheral artery disease. 4–11 Thus, measurement of circulating ox-LDL level may be useful not only for predicting future cardiovascular events, but also for identifying the factors that could determine LDL susceptibility to oxidation, which would provide us variable information to decide a therapeutic strategy for the treatment of high-risk patients.
Several groups have reported specific enzyme-linked immunosorbent assay (ELISA) systems to measure plasma levels of ox-LDLs. 12–14 However, there are some limitations in applying their assays for a clinical study as follows: (i) when blood samples are to be assayed based on these methods, the LDL fraction has to be obtained by ultracentrifugation of each sample, which could make the assays time-consuming and laborious, and (ii) since the previous methods determined circulating ox-LDL levels using monoclonal antibodies specific to aldehyde- or oxidized phosphatidylcholine-modified LDLs, their assays could not detect other forms of circulating ox-LDLs in the plasma; in other words, the assays could reflect only minor parts of ox-LDLs in human plasma. 12–14 Further, although other analytical methods such as NMR spectroscopy, capillary electrophoresis, agarose electrophoresis and high-performance liquid chromatography (HPLC) were introduced to identify LDLs in human serum, 15–18 these assays were not necessarily specific for detecting oxidized forms of LDLs or time-consuming ones. We have previously developed a convenient method for fractionating lipoproteins by HPLC with a diethylaminoethyl (DEAE)-glucomannan column. 19,20 Plasma concentrations of HDL-cholesterol (HDL-C), LDL-cholesterol (LDL-C), very-low-density lipoprotein-cholesterol (VLDL-C) and chylomicron determined by this method were highly correlated with those estimated by an ultracentrifugation method. 19,20 Therefore, in this study, we developed a rapid, convenient and reliable method for evaluating ox-LDLs systemically in human plasma using a novel anion-exchange HPLC (AE-HPLC) procedure with postcolumn reaction.
Materials and methods
Reagents
Cholesterol oxidase (22 U/mg) from Streptomyces sp., peroxidase (150 U/mg) from horseradish and cholesterol esterase (122 U/mg) from Pseudomonas sp. were purchased from Toyobo (Osaka, Japan). Homovanillic acid was from Sigma-Aldrich (Tokyo, Japan). Triton X-100 was obtained from Dojindo Laboratories (Tokyo, Japan). Specific anti-apolipoprotein B100 (apoB100) antibodies were obtained from Funakoshi Co Ltd (Tokyo, Japan), while anti-malondialdehyde (MDA), anti-4-hydroxy-2-nonenal (4-HNE) and anti-hexanoyl-lysine adduct (HEL) antibodies were obtained from Nikken Soil Co Ltd (Shizuoka, Japan). A horseradish peroxidase-conjugated goat anti-mouse secondary antibody was obtained from DAKO Cytomation (Kyoto, Japan). All other chemicals of analytical reagent grade were from Wako Pure Chemicals Co Ltd (Osaka, Japan).
Subjects
Control blood samples were collected from six healthy volunteers (3 men and 3 women, age: 41.8 ± 8.1 y) after 12-h fasting, and then stored at −20°C until analysis. Fractionated components of VLDL (0.930 < d < 1.006), LDL (1.019 < d < 1.063) and HDL (1.063 < d < 1.210) were obtained from the plasma by ultracentrifugation as described previously. 21 Then, plasma or lipoproteins were separated by HPLC. In addition, plasma samples were also obtained from 61 healthy subjects to evaluate LDL particle size in each LDL fraction. Written informed consent was obtained from all participants in this study.
HPLC methods
Our AE-HPLC system CLASS LC-10A (Shimadzu Corporation, Kyoto, Japan) is shown in Figure 1a. It consisted of four LC-10A pumps for delivering the two eluents, the cholesterol reagent and the alkaline reagent, an SIL-10A autosample injector with a sample cooler, two CTO-10AC column ovens and an RF-10AXL fluorescence detector. We developed a new type of anion-exchange column (MCI® GEL ProtEx-DEAE purchased from Mitsubishi Chemical Co, Tokyo, Japan), with some modifications of commercially available MCI® ProtEx DEAE (Mitsubishi Chemical Co). Eluent A was 20 mmol/L sodium phosphate buffer, pH 7.0, containing 1 mmol/L EDTA-3Na, and eluent B was 20 mmol/L sodium phosphate buffer, pH 7.0, containing 0.5 mmol/L NaCl and 1 mmol/L EDTA-3Na. Plasma and serum samples were diluted 10-fold with phosphate-buffered saline and 20 μL aliquot of the sample was injected into the HPLC system. Plasma or lipoproteins were separated by step gradient elution as shown in Figure 1b. The separation was carried out at 25°C at a flow rate of 1.0 mL/min.
Schematic diagram of our anion-exchange high-performance liquid chromatography system (a) and the conditions for the stepwise elution used in this study (b)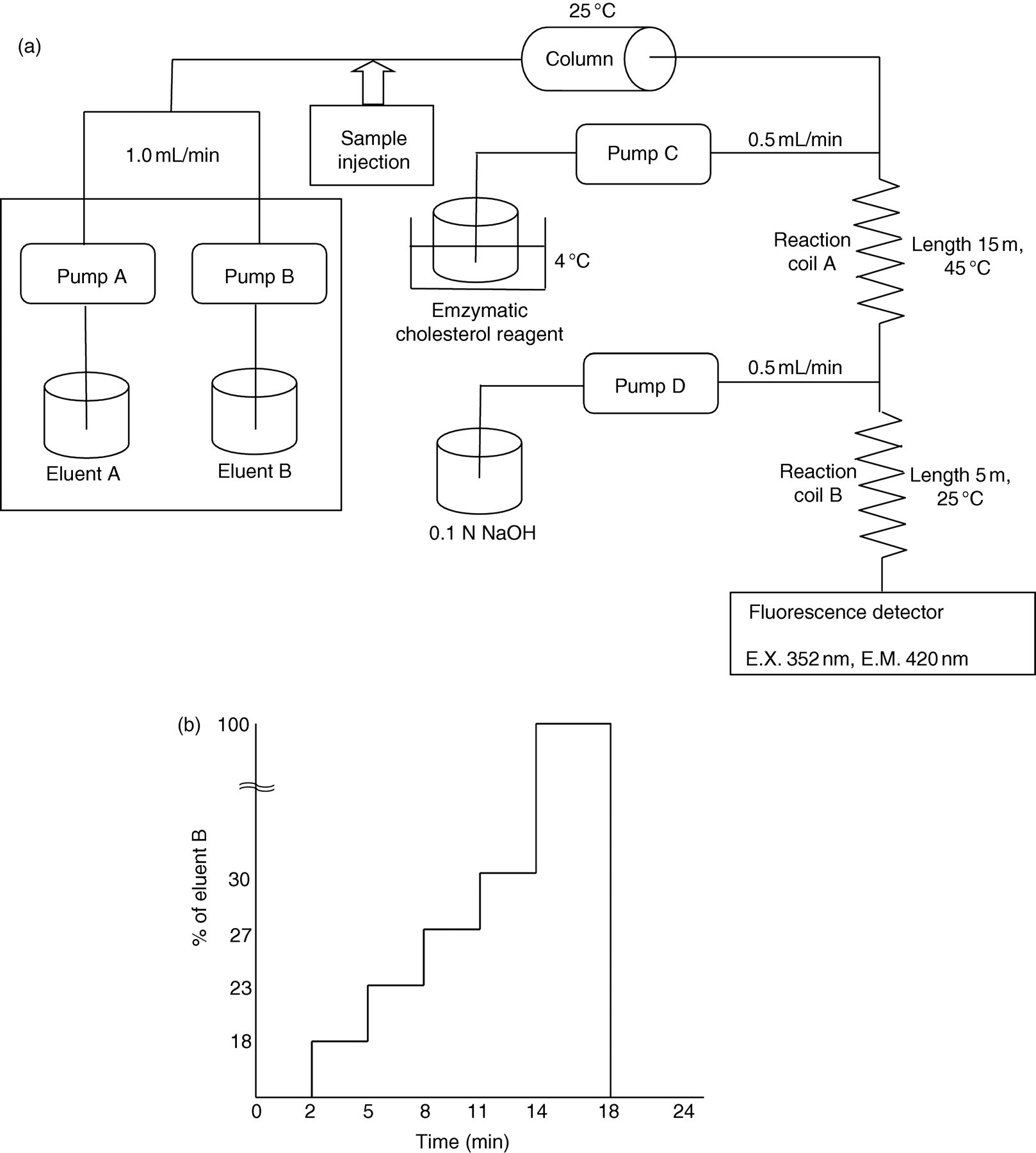
A postcolumn reaction was performed to determine cholesterol levels in each lipoprotein fraction. In brief, cholesterol esterase (122 U/mL, 0.5 mg/L), cholesterol oxidase (22 U/mL, 1.0 mg/L), peroxidase (150 U/mL, 10 mg/L) and homovanillic acid (50 mg/L) were dissolved in 20 mmol/L sodium phosphate buffer, pH 7.0, containing 0.2% Triton X-100 (referred to as cholesterol reagent), and delivered into the reaction coil at a flow rate of 0.5 mL/min. A Teflon coil (15 m × 0.5 mm I.D.) was employed for the enzymatic reaction and placed in a column oven (45°C). The 0.1 mmol/L NaOH solution was delivered into a stainless-steel reaction coil (0.5 m × 0.5 mm I.D.) at a flow rate of 0.5 mL/min to render the effluent alkaline. Cholesterol levels in each lipoprotein fraction were measured at an excitation wavelength of 325 nm and an emission wavelength of 420 nm using a fluorescence detector. Chromatographic data were collected with a CBA-10A interface, transmitted to a personal computer and integrated using the CLASS-LC10 software (version 1.41).
Oxidation of LDLs in vitro
LDLs were obtained from the plasma by ultracentrifugation and then oxidized in vitro with 5 μmol/L CuSO4 or 2 mmol/L 2,2-azo-bis (2-amidinopropane)-2HCl (AAPH), a temperature-dependent free radical generator, at 37°C for 0, 0.5, 1, 2, 4, 6 or 8 h, respectively. 22,23
Preparation of aldehyde-modified LDLs
LDLs were isolated from the plasma by ultracentrifugation, dialyzed against 0.01 mmol/L sodium phosphate buffer, pH 7.4, containing 0.15 mmol/L NaCl and 0.01% EDTA (buffer A), and then coupled with MDA, n-butyraldehyde (BUA), propionaldehyde (PPA) and n-valeraldehyde (VLA) for three hours at 37°C, respectively, to obtain various types of aldehyde-modified LDLs according to the method of Watson et al. 24
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis and Western blot analysis
LDLs isolated from the plasma by ultracentrifugation, LDL-1, LDL-2 and LDL-3 subfractions were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto Immobilon-PSQ membrane (Millipore, MA, USA), as described previously. 25 Membranes were probed with specific antibodies raised against apoB100, MDA, 4-HNE or HEL at 4°C overnight, and then with alkaline phosphatase-conjugated goat anti-mouse antibodies. Immune complexes were visualized with an enhanced chemiluminescence detection system (Moss Inc, Baltimore, MD, USA).
Measurement of LDL particle size
LDL particle size was determined by measuring the migration distance of each LDL fraction with non-denaturing PAGE according to the method described previously. 8
Statistical analysis
All values are presented as the mean ± standard deviation. Statistical significance was evaluated using non-parametric Mann–Whitney's U test (GraphPad InStat Ver. 3.06), and P < 0.05 was considered statistically significant.
Results
Separation and identification of LDL subfractions
Figure 2 shows the typical chromatogram peaks of lipoproteins in whole plasma by HPLC (solid line). As shown in Figure 2, lipoproteins were separated into five peaks, HDL, three subfractions of LDLs (designated LDL-1, LDL-2 and LDL-3 by their elution order) and VLDL within 24 min using our HPLC system without extraction of the lipoproteins by ultracentrifugation prior to the assay. The chromatogram was essentially the same as that observed with lipoproteins obtained by ultracentrifugation (bold line). The ratio of LDL-1, LDL-2 and LDL-3 to total LDLs were 72.7 ± 2.8%, 23.7 ± 2.7% and 3.5 ± 0.8%, respectively. We also confirmed that apoAI and apoAII were detected only in HDL fractions separated by HPLC, while apoB was detected in LDL and VLDL fractions (data not shown). Within- and between-day assay coefficients of variation of our HPLC method was <0.7% and <2.4% for LDL-1, <2.6% and <4.4% for LDL-2 and <2.2% and <1.7% for LDL-3, respectively. Serum samples were serially diluted ten-fold with phosphate-buffered saline. The assay linearity was shown intact with the serial dilution (coefficients of variation <5%). Freezing and thawing of the samples (at −20°C) were repeatedly performed at the stored days of 1, 7, 14 and 28. As shown in Table 1, four cycles of freezing and thawing gave no effects on the assay results. Further, 12-month storage of samples also did not affect the assay results. However, the levels of LDL-2 and LDL-3 decreased up to about 20% when the samples were stored at 4°C for seven days.
Typical chromatograms of plasma (solid line) and LDLs isolated from the plasma by ultracentrifugation (bold line). LDL, low-density lipoprotein Effects of freezing and thawing on LDL subfraction values LDL, low-density lipoprotein Data are shown as the percentage of the values obtained without freezing and thawing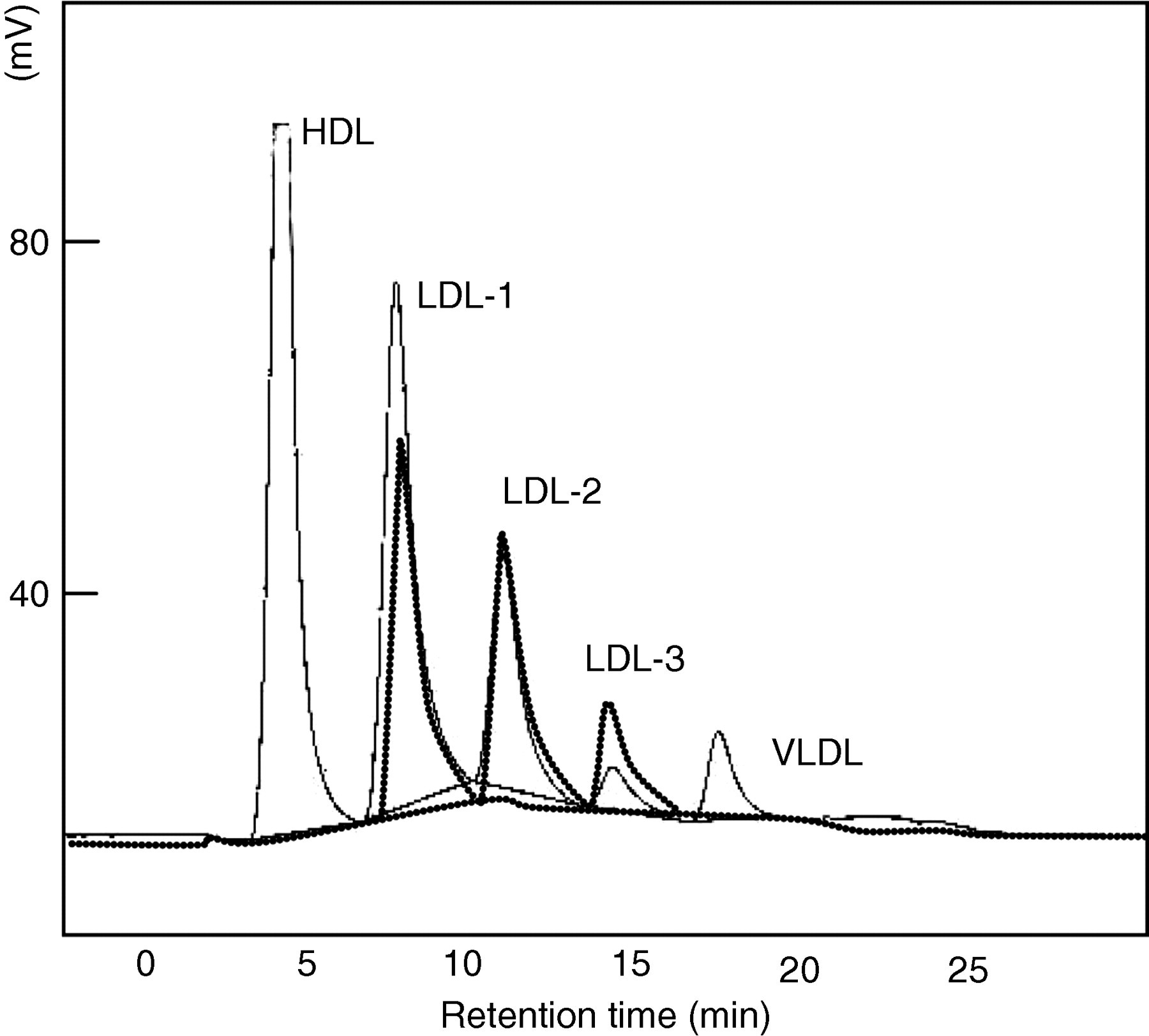
Characterization of LDL subfractions
Western blotting analysis revealed that apoB100 in LDL-2 and LDL-3 but not LDL-1 was fragmented (Figure 3a). Further, various types of aldehyde-modified LDLs, such as MDA-LDL, 4-HNE-LDL and HEL-LDL, were detected only in small particles of apoB100 in LDL-2 and LDL-3, especially in LDL-3 (Figure 3b–d, arrowheads).
Western blotting analysis of LDLs isolated from the plasma by ultracentrifugation (lane 1), LDL-1 (lane 2), LDL-2 (lane 3) and LDL-3 fractions (lane 4). Each sample was reacted with anti-apoB100 (a), anti-MDA (b), anti-4-HNE (c) and anti-HEL (d) antibodies. LDL, low-density lipoprotein; apoB100, apolipoprotein B100; MDA, malondialdehyde; 4-HNE, anti-4-hydroxy-2-nonenal; HEL, hexanoyl-lysine adduct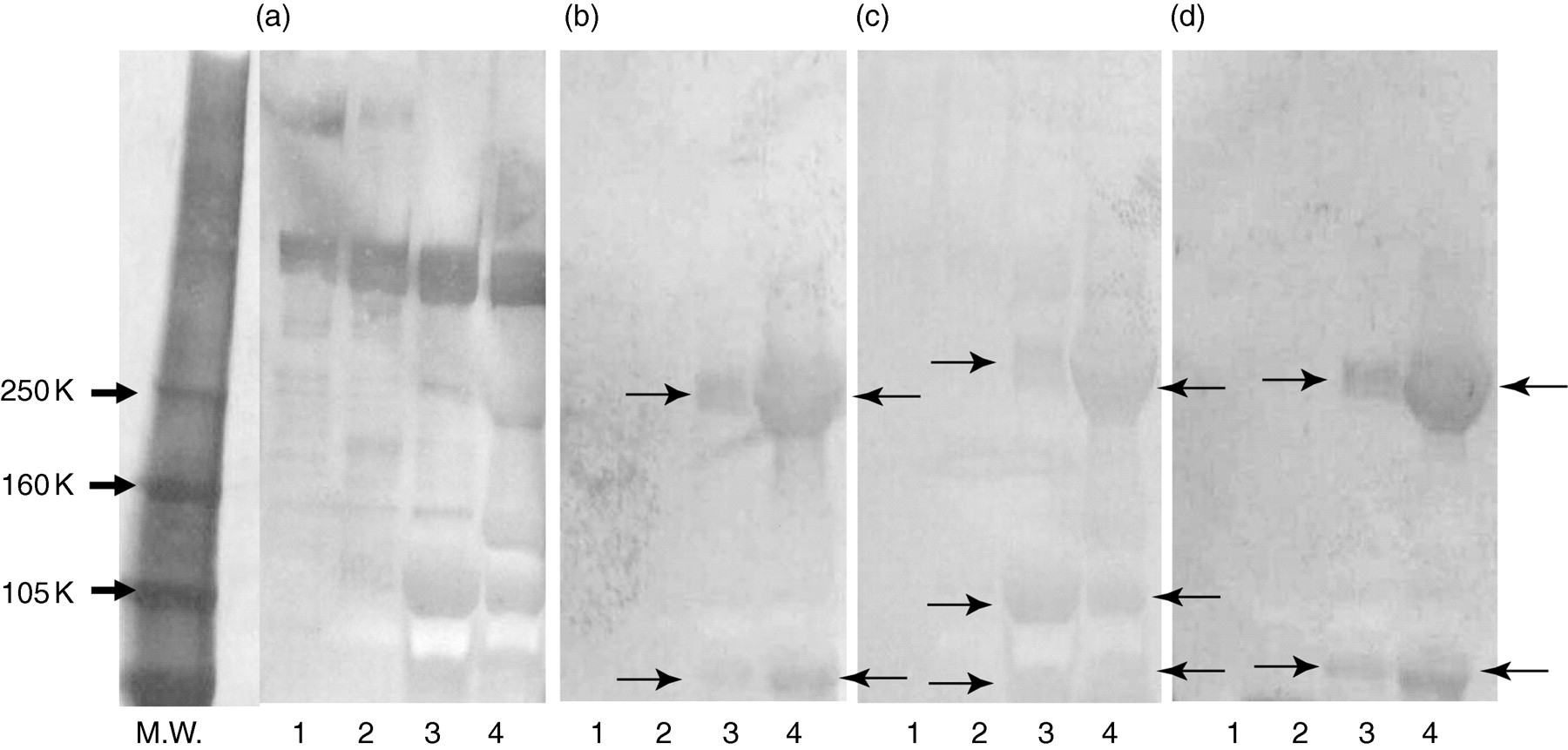
LDL fraction patterns of in vitro-prepared ox-LDLs
LDL, low-density lipoprotein; ox-LDL, oxidized-LDL; LDL-C, LDL-cholesterol; AAPH, 2,2-azo-bis (2-amidinopropane)-2HCl
LDL fraction patterns of various aldehyde-modified LDLs
LDL, low-density lipoprotein; LDL-C, LDL-cholesterol; BUA, n-butyraldehyde; PPA, propionaldehyde; n-valeraldehyde, MDA, malondialdehyde
Particle size of each LDL fraction
As shown in Figure 4, the particle size was found to correlate with the ratio of LDL-1 to total LDL-C (r = 0.83) in 61 healthy subjects (30 men, age: 41.7 ± 6.4 y; and 31 women, age: 39.2 ± 7.0 y), while it was inversely associated with the ratio of LDL-2 (r = −0.84) and LDL-3 to total LDL-C (r = −0.70).
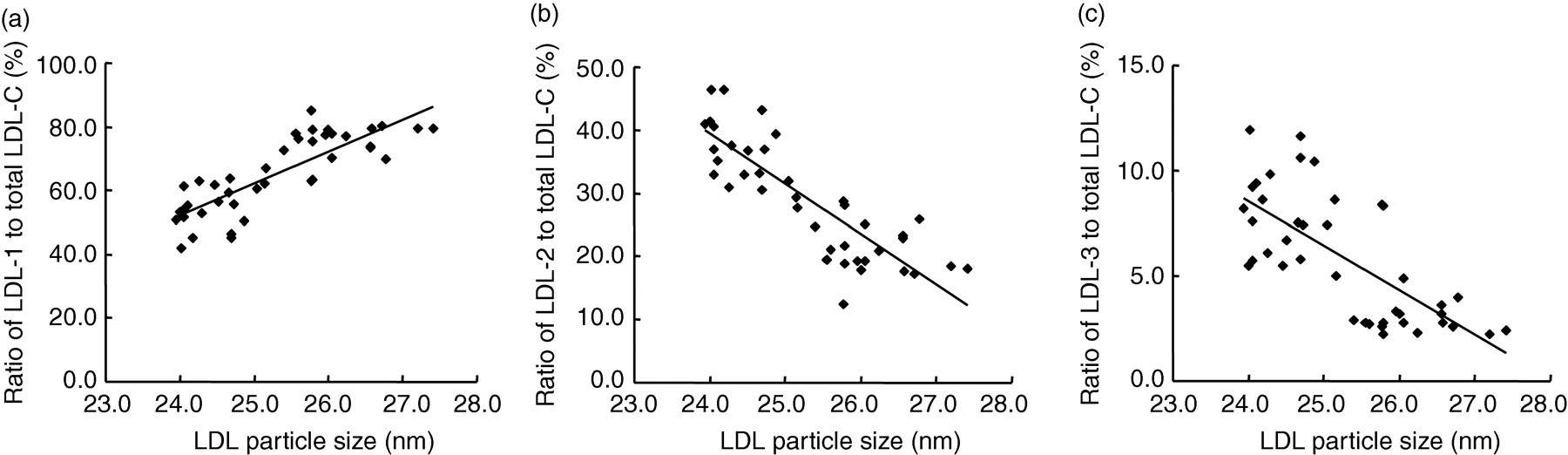
LDL particle sizes of each LDL fraction. Correlation of the ratio of each LDL fraction to total LDL with particle sizes was shown. LDL, low-density lipoprotein
Discussion
Several ELISA systems have been reported for measuring ox-LDLs in human plasma. 12–14 However, their systems could only detect specific types of ox-LDLs, such as aldehyde- or oxidized phosphatidylcholine-modified LDLs, and to isolate LDLs from the plasma by ultracentrifugation prior to the assays was time-consuming. In order to overcome these limitations, we employed here a modified anion-exchange HPLC method for measuring ox-LDL levels in human plasma. This method was useful and timesaving because (1) LDL subfractions were completely separated within 24 min without prior lipoprotein extraction and (2) it can easily permit quantitative determination of all forms of ox-LDLs present in the circulation.
Our HPLC method separated LDLs into three fractions (designated as LDL-1, LDL-2 and LDL-3 by their elution order). We identified LDL-3, the most strongly retained fraction as ox-LDLs on the basis of the following reasons: (1) agarose gel electrophoresis revealed that LDL-3 run fastest in the anodic direction, thus reflecting their negative net charges on the molecules; (2) apoB100 in LDL-3 was most fragmented, and antibodies directed against lipid peroxidation products, such as MDA, 4-HNE or HEL, reacted to apoB100 in LDL-3, but not LDL-1 or LDL-2; (3) when LDLs were isolated from the plasma by ultracentrifugation and then oxidized in vitro by copper sulphate or AAPH for more than eight hours, all of the LDLs migrated at the position of LDL-3; and (4) various types of aldehyde-modified LDLs, such as MDA-LDL, BUA-LDL, PPA-LDL and VLA-LDL, were also separated at the position of LDL-3, but not LDL-1 or LDL-2 on HPLC analysis. Further, our HPLC method was accurate and reproducible. Four cycles of freezing and thawing and 12-month storage of samples gave no effects on the assay results. It is well known that ox-LDLs are more negatively charged than native LDLs and show small, dense characteristics due to oxidative fragmentation of apoB100 of the LDL molecule, 26 further supporting the reliability of our assay.
The increased prevalence of small, dense LDL particles has been reported to correlate with atherosclerotic diseases in humans. 22 LDL particles are heterogeneous with respect to size and density of lipid composition, with small, dense LDL particles being highly atherogenic as a result of their increased ability to penetrate into the arterial wall and also their lower binding affinity for the LDL receptor, prolonged plasma half-life and lower resistance to oxidative stress. 26 Therefore, measuring LDL particle size by gradient gel electrophoresis or analytical centrifugation is clinically useful to identify high-risk patients for cardiovascular disease. However, the methods are too laborious for general clinical practice. In this study, using our HPLC method, LDL particle size was found to correlate positively with the ratio of LDL-1 to total LDL-C in healthy subjects, while it was inversely associated with the ratio of LDL-2 and LDL-3 to total LDL-C. These observations suggest that our HPLC method may also be convenient and reliable for detecting small, dense and oxidized modified LDL fractions in human plasma.
We have previously found that LDL-3 fractions not only induce apoptotic cell death, but also stimulate monocyte chemoattractant protein-1 production in cultured human umbilical vein endothelial cells, both of which are blocked by the treatment with an anti-oxidant N-acetycysteine. 27 Further, our recent study has shown that LDL-3 levels are strongly associated with the metabolic syndrome in apparently healthy subjects 28 and with prevalence of silent lacunar infarction in patients with essential hypertension. 29 Therefore, our HPLC method for measuring plasma levels of ox-LDL may be useful to evaluate the efficacy of novel therapeutics on atherosclerosis-related disorders.
Limitations
In this paper, ox-LDL samples were prepared in vitro by incubating LDLs with 5 μmol/L CuSO4 or 2 mmol/L AAPH, according to the methods as described previously. 22,23 Therefore, we did not know how extensively LDLs were modified in the present study. So, it would be helpful to determine the oxidation levels of modified LDLs used here by conjugated diens, thiobarbituric acid-reactive substances or relative electrophoresis mobility. Since ox-LDLs were present in LDL-2 fraction, albeit lesser concentrations than in LDL-3 subfraction, there is some limitation in quantitative determination of ox-LDLs by our method.
DECLARATIONS
Footnotes
Acknowledgements