
Abstract
Background
The clinical requirements of the users of assay results must be at the centre of assay development. We aimed to develop a single liquid chromatography tandem mass spectrometry (LC-MS/MS) assay for drugs of abuse in urine that would meet the needs of our service users and replace the multiple screening and confirmatory techniques previously in use.
Methods
After discussion with our users, it was decided that 13 drugs and metabolites should be measured in our panel: morphine, codeine, norcodeine, dihydrocodeine, 6-monoacetylmorphine, acetyl codeine, methadone and its metabolite, buprenorphine and its metabolite, amphetamine, benzoylecgonine and cotinine. Urine samples were prepared by the addition of internal standard, enzymatic hydrolysis and solid-phase extraction. Chromatography conditions were optimized so that the analytes were separated within a run time of 6 min. Optimal parent to daughter m/z ion transitions were chosen for all drugs and daughter ion ratios were used.
Results
The LC-MS/MS assay was successfully validated with acceptable precision and lower limits of quantification for all drugs. No matrix effects were seen. The results produced by the LC-MS/MS assay compared well with the previous combination of techniques in use.
Conclusions
We have developed and validated a fit-for-purpose LC-MS/MS assay for 13 drugs of abuse in urine that obviates the need for multiple screening and confirmatory analytical techniques.
Introduction
Addiction to illicit drugs has many detrimental effects on individuals and society as a whole. The aims of drug treatment programmes are eventual abstinence with concomitant reduction in harmful behaviour. Drug screening by laboratories plays a vital role in these programmes. The results are used to ensure that clients are compliant with prescribed substitution therapies and to monitor illicit drug use, thus enabling an honest relationship to be built between a client and his/her key worker. The UK Department of Health published a 10-year drug strategy in 2008. 1 One of its key aims is for drug treatment services to become more personalized towards meeting an individual's needs, an aim which can only be achieved with comprehensive and accurate drug screening.
The laboratory provides a regional screening service for drugs of abuse in urine samples for the local mental health trust, supplying us with over 26,000 samples per year. This workload requires quick, robust methods of analysis which are suitable for handling a large proportion of positive results. This is in contrast to laboratories that handle large numbers of samples for pre-employment screening where the proportion of positive samples is much lower. 2
Previously in our laboratory, as in many other laboratories, 3,4 urine samples were analysed for drugs of abuse using multiple screening and confirmatory stages (Table 1). The results from the different methods were combined and interpreted to produce a report that stated which drugs had been detected.
Previous combinations of methods used for drug screening
Together, this combination of techniques produced accurate and reliable results. However, the use of multiple steps was inefficient and slow as each urine sample was repeatedly tested by different techniques. Of these previous methods, no one method alone would be suitable for use without the others. Immunoassays are easily automated and simple to perform but problems with non-specificity, in particular with amphetamine group immunoassay, are well recognized. 5 High-performance thin layer chromatography (HPTLC) is more specific but can be insensitive and requires expertise and experience for interpretation. This can be a particular difficulty in a busy clinical laboratory where staff may only work in different sections on a rotational basis. Gas chromatography mass spectrometry (GC-MS) is a highly specific technique, but can require complicated sample preparation if derivatization is required and can have long analysis time and difficult interpretation.
The use of liquid chromatography tandem mass spectrometry (LC-MS/MS) techniques in drugs of abuse testing is increasing. 6 It is recognized as a sensitive and specific technique and with rapid analysis times can obviate the need for expensive pre-screening of samples by immunoassay. We aimed to develop and validate a single LC-MS/MS assay that could report a number of different drugs in urine as either ‘detected’ or ‘not detected’ when compared with a cut-off concentration. The new single LC-MS/MS assay would replace all of our previous methods so that multiple analyses on the same sample would no longer be necessary.
Changing our approach to drugs of abuse analysis provided us an opportunity for liaison with our clinical service users. An initial meeting was held between the members of the laboratory and the Mental Health Trust consultants to decide which drugs should be measured in the new single LC-MS/MS screen (Table 2). It is known that abused drugs differ with locality. 7,8 Collaboration with our service users thus enabled the new assay to meet all of their clinical requirements without wasting resources measuring drugs that they have no need of.
The 13 drugs and metabolites aimed to be included in our single LC-MS/MS assay. This panel was a joint decision between laboratory and mental health trust workers
Materials and methods
Standards and controls
Stock drugs standards in methanol (Cerilliant, obtained from LGC Promochem, Middlesex, UK) were diluted in water to produce a combined standard with all drugs present at their cut-off concentration. UKNEQAS (United Kingdom National External Quality Assessment Service) clinical cut-off concentrations were used, or concentrations obtained through literature searches where UKNEQAS concentrations were not available (Table 3). 11,12 Controls were prepared in drug-free urine at concentrations just above and below the cut-off concentration (Table 3).
Concentration of drugs present in combined standard and controls
Five internal standards were used in the analysis, morphine D3, methadone D9, benzoylecgonine D3, amphetamine D5 and EDDP D5 (Cerilliant, obtained from LGC Promochem, Middlesex, UK). Stock solutions were diluted in water to produce a combined internal standard with all deuterated compounds present at 1000 μg/L.
Chromatography
Chromatography was performed on an Acquity UPLC® system with a HSS T3 1.8 μm 2.1 × 50 mm column (both from Waters, Milford, USA). Mobile phase A consisted of 5 mmol/L ammonium formate with 0.05% formic acid (both from VWR, Leicestershire, UK) in 18 MΩ water. Mobile phase B was LC-MS grade methanol (Fisher Scientific, Loughborough, UK). A flow rate of 0.35 mL/min was maintained for the run time of 6 min. The flow was diverted to waste until 1.5 min and then again after 5 min. In between these times there was a linear gradient of mobile phase B from 30 to 60% to provide adequate separation of the analytes. The injection volume from the autosampler was 7.5 μL.
Mass spectrometry
Mass spectrometric detection was performed using a TQ Detector (tandem quadrupole mass spectrometer) (Waters, Milford, USA). Electrospray positive mode was used with a source temperature of 120°C, a desolvation temperature of 350°C, a cone gas flow of 50 L/h and a desolvation gas flow of 800 L/h. Transitions for each drug of interest were chosen by tuning the instrument with 1000 μg/L solutions of each drug in 70:30 mobile phases A:B. The cone voltage and collision energy were optimized for each drug (Table 4). TargetLynx™ software (Waters, Milford, USA) was used to integrate and quantify all drugs in the standards, quality controls and samples.
Optimal transitions, dwell times, cone voltages and collision energies for each drug
Sample preparation
To 1 mL urine standard control, 200 μL combined internal standard (1000 μg/L) was added. This was incubated with β-glucuronidase from Helix pomatia (Sigma-Aldrich, Dorset, UK) and a pH 5 sodium acetate buffer at 56°C for 1 h to allow hydrolysis of the glucuronide-drug conjugates and increase the yield of free drugs.
After the addition of a phosphate buffer at pH 6 and centrifugation at 3000 rpm (rotations per minute) for 15 min, the samples underwent solid phase extraction using Strata Screen C 3 mL cartridges (Phenomenex, Cheshire, UK). After the preparation of the columns, the sample was added, cleaned and then eluted using dichloromethane with propan-2-ol (both from VWR, Leicestershire, UK) and ammonia (Fisher Scientific, Loughborough, UK) in the proportion 75:23:2, respectively. The samples were evaporated to dryness and then re-constituted in 25% aqueous methanol before they were transferred to a 96-well plate that was heat sealed and placed on the autosampler.
Method validation
The method was validated in the following ways:
Matrix effects: An extracted blank water sample, three different extracted drug-free urine samples and three urine samples diluted 50:50 with methanol were run against a constant background infusion at 40 μL/min of a solution containing all 13 drugs at 2 mg/L. The traces produced by the water and urine samples were compared to look for a decrease or increase in signal at the relevant retention times. Specificity: Since some opiates and metabolites have the same molecular weight, previously described interferences were investigated. A blank urine was spiked separately with 2000 μg/L hydrocodone and 2000 μg/L hydromorphone. The samples were then analysed to look for the presence of codeine (which has the same nominal molecular weight as hydrocodone, 299) and morphine and norcodeine (which both have the same nominal molecular weight as hydromorphone, 285). Precision: Both the positive and negative controls were run 10 times within one batch and in 10 separate batches and the corresponding coefficient of variance (CV) values calculated to determine within and between batch precision around the cut-off concentration. Lower limit of quantification: Repeated dilutions of the negative control were done with blank urine and each diluted solution measured 10 times to determine the concentration of each drug at which the CV became greater than 15%. Linearity: Samples that contained very high levels of amphetamine, benzoylecgonine and morphine were diluted with blank urine in different ratios. The concentrations measured in the resulting samples were plotted to determine at what concentration the response became non-linear. Carryover: Samples containing high levels of amphetamine, benzoylecgonine and morphine were measured before the measurement of several blank water samples in order to determine the extent of any carryover in the system. Method comparison: Over 1000 samples that previously had a routine drug analysis performed for opiates, methadone and EDDP, benzoylecgonine and amphetamine by the original analytical techniques (Table 1) were re-analysed by the new LC-MS/MS combined method. We compared the overall final report generated by our old combination of methods (‘detected’ or ‘not detected’) with the result determined by the new method (‘detected’ or ‘not detected’, when compared with a cut-off concentration). For buprenorphine and norbuprenorphine, we compared over 100 samples that had been measured by our previous GC-MS method.
Results
Optimal transitions, cone voltages and collision energies were found for each drug and internal standard by tuning (Tables 4 and 5). Where two daughter ions were used, the first was used as a quantifier ion and the second was used as a qualifier ion. The ion ratio is the ratio of the quantifier daughter ion response to the qualifier daughter ion response and was used to increase the confidence of identification. An ion ratio within ±25% of the expected value was considered acceptable when processing the results. 13
Optimal transitions, dwell times, cone voltages and collision energies for each internal standard
The liquid chromatography was optimized to ensure suitable separation of the analytes (Figure 1). This separation allowed the mass spectrometric detection to be split into different time windows based on analyte retention time. This ensured that sensitivity was not compromised by the simultaneous detection of multiple analytes.
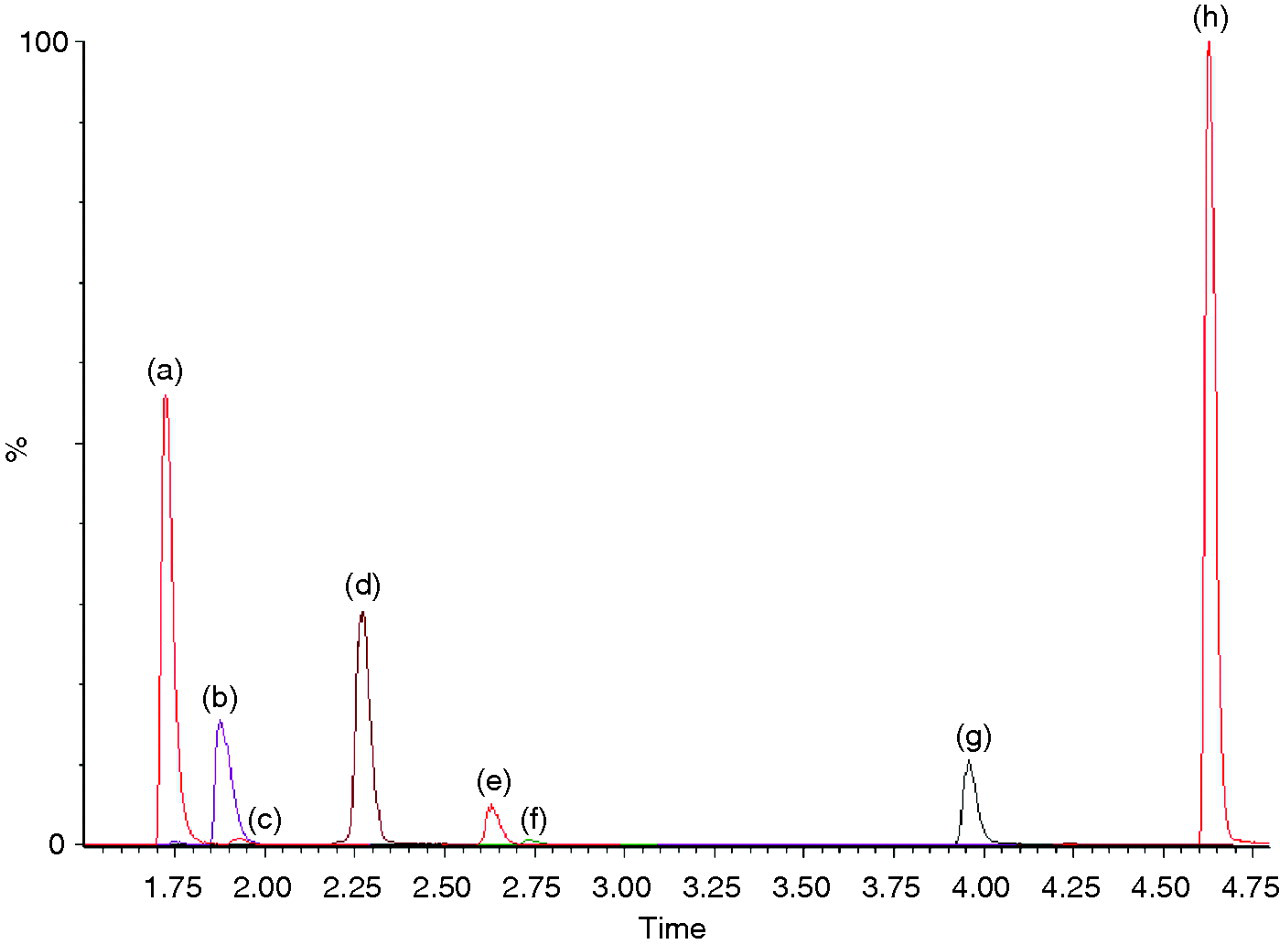
Chromatogram of positive control (a) morphine and cotinine, (b) codeine and dihydrocodeine (DHC), (c) norcodeine, (d) amphetamine, (e) benzoyl ecgonine, (f) acetyl codeine, (g) EDDP and (h) methadone. 6-MAM (retention time 2.1 min), norbuprenorphine (retention time 3.7 min) and buprenorphine (retention time 4.3 min) cannot be seen due to their presence at low concentration in the positive control
It was found that deuterated compounds could only be used as internal standards for their non-deuterated equivalent only. As an example, the amount of morphine D3 measured was reduced in a sample containing a very high concentration of morphine. If morphine D3 was then also used as an internal standard for a second opiate, the concentration of this second opiate would be falsely increased.
The method was fully validated before incorporation into routine use.
Matrix effects: The traces produced by three prepared urines (by both solid-phase extraction and dilution with methanol), when a high concentration of each drug was infused, was compared with that similarly produced by an extracted blank water sample. For all drugs, no significant difference in signal between blank water and each of the three extracted urine samples was seen at the relevant retention times (results for methadone and one urine sample shown, Figure 2). However, the urine samples solely diluted with methanol did show a reduced signal and areas of ion suppression due to matrix effects. Specificity: Although a peak due to 2000 μg/L hydrocodone was seen in the codeine transitions when no codeine was present, it had a different retention time to that of codeine (1.98 min as opposed to 1.88 min) and also a different ion ratio (0.5 as opposed to 1.9). No significant peaks were seen in the norcodeine transitions due to the presence of 2000 μg/L hydromorphone. A peak due to hydromorphone was seen in the morphine channel and although it had only a slightly different retention time to morphine (1.78 min as opposed to 1.75 min) the ion ratio was very different (0.08 as opposed to 0.93). Precision: The positive and negative controls showed acceptable precision when run both within and between batches (Table 6). Lower limit of quantification: With the exception of -6-monoacetyl morphine (6-MAM), the lower limit of quantification was demonstrated to be below the cut-off concentration for all drugs (Table 6). For 6-MAM, 10 μg/L was the lower limit of quantification and also the cut-off concentration. Therefore, the 6-MAM negative control and standard both contained the same concentration. Linearity: Amphetamine, morphine and benzoylecgonine were linear until concentrations were greater than 40, 100 and 250 times their cut-off concentrations, respectively. When non-linearity did occur the response flattened rather than decreased. Carryover: Less than 0.1% carryover was observed following highly concentrated (>100,000 μg/L) amphetamine, morphine and benzoylecgonine samples. Method comparison: The samples used in the comparison studies consisted of both positive and negative results for each drug. The new single LC-MS/MS method results compared well with the results produced by our old combination of methods (Table 7).
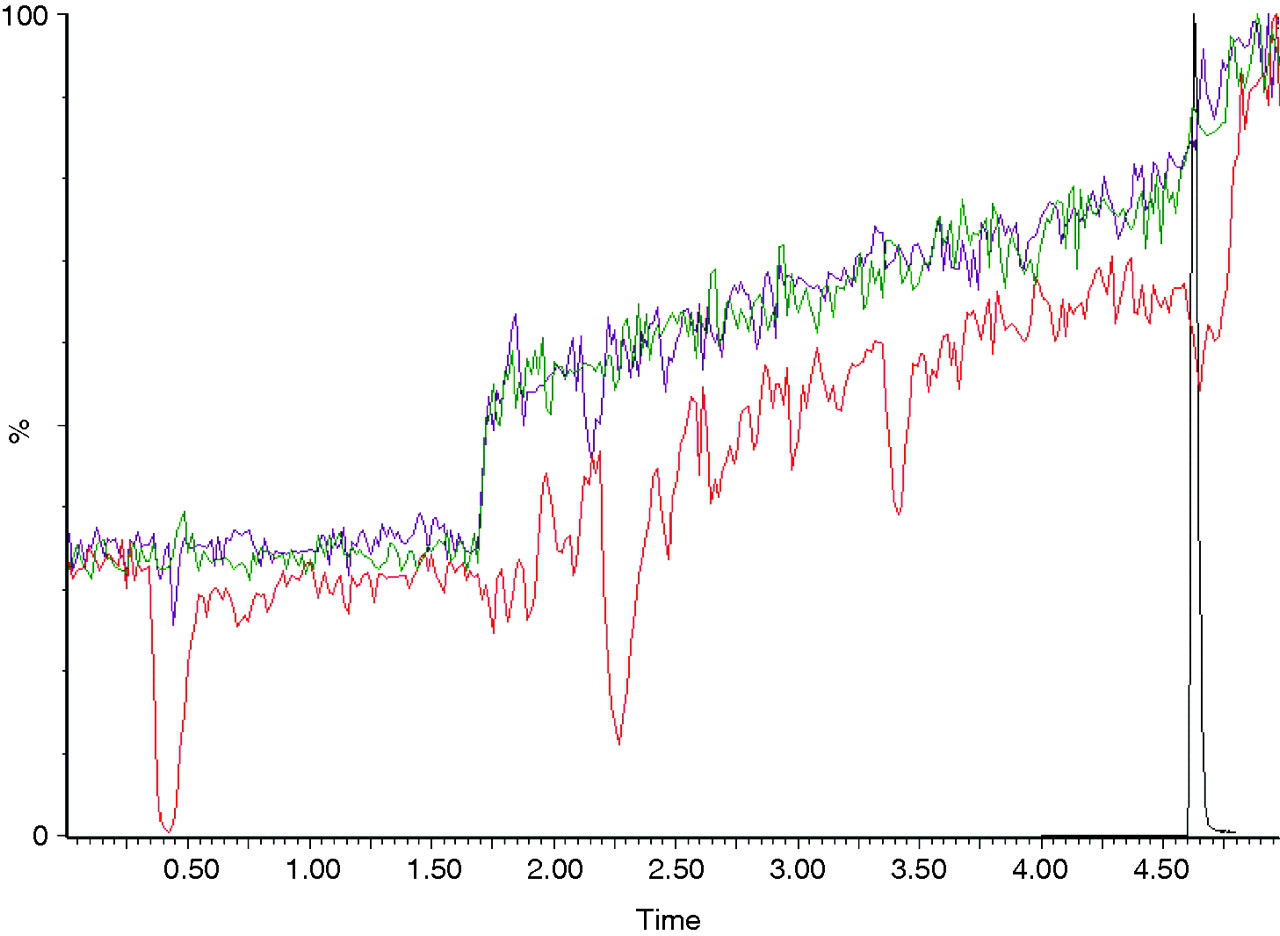
Traces produced in the methadone primary transition by an extracted blank water sample (green line), an extracted urine sample (purple line) and a urine sample diluted 50:50 with methanol (red line) all with a concomitant constant infusion of 2 mg/L methadone. The methadone retention time is overlaid (black line). Similar traces were produced for all drugs with three different urine samples
Within batch precision, between batch precision and lower limit of quantitation for all drugs
LLoQ, lower limit of quantitation
Results of the comparison between the old combination of methods and the new single LC-MS/MS method
Discussion
We have developed and validated a single LC-MS/MS assay for 13 drugs of abuse and metabolites to meet the requirements of our service users.
Optimal transitions for each drug were identified. For the majority of drugs, two separate ion transitions were used to increase the confidence of identification and allow the use of ion ratios. Tandem quadrupole mass spectrometers are undoubtedly highly specific instruments. However, it is important to be aware that non-specificity can still occur, particularly in drugs of abuse analysis where many drugs and metabolites have the same nominal molecular weight and similar structures. We anticipated this problem with morphine and norcodeine as both have a nominal molecular weight of 285 and differ structurally only in the position of a methyl group (Figure 3). The potential for mis-identification was greatly reduced by attempting to choose daughter ions that were unique to each molecule, chromatographic separation and the use of different ion ratios. These factors were also useful in distinguishing hydrocodone from codeine and hydromorphone from morphine or norcodeine.
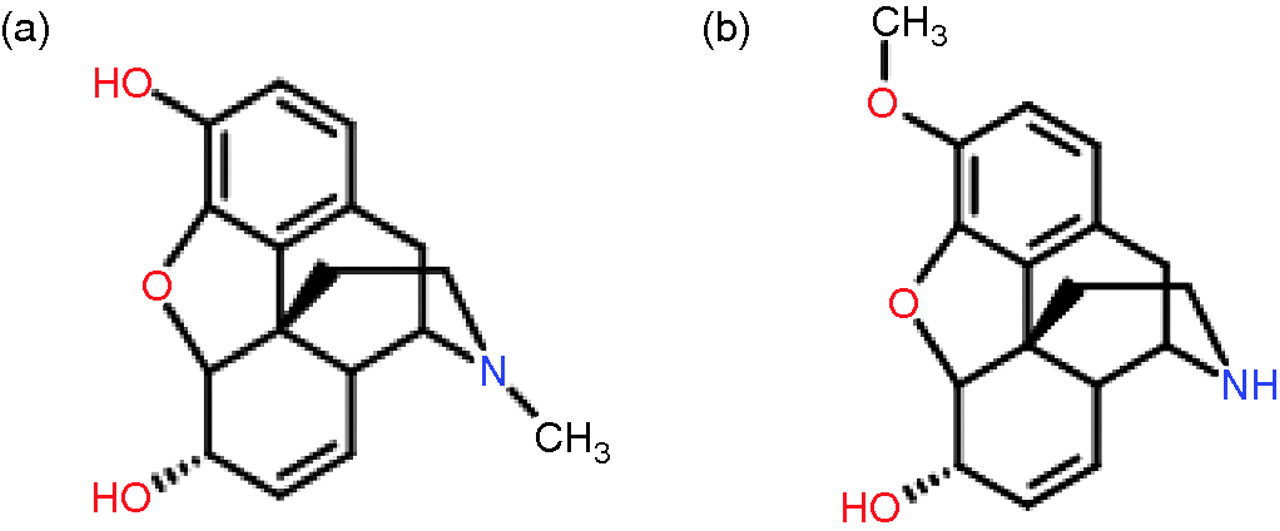
Molecular structure of (a) morphine and (b) norcodeine
For buprenrorphine and norbuprenorphine, only one ion transition was used for each compound. It was difficult to achieve adequate sensitivity for these two analytes and the use of two transitions for each, further decreased the sensitivity. Since both buprenorphine and norbuprenorphine are large molecules (molecular weight 467 and 413, respectively), the possibility of mis-identification is lower and so use of only one daughter ion was considered acceptable. Due to a lack of sensitivity for buprenorphine and norbuprenorphine, we could not report to the UKNEQAS recommended cut-off of 5 μg/L. 11 Our experience with GC-MS analysis of buprenorphine and norbuprenorphine suggested that clients taking regularly prescribed buprenorphine would typically have values much higher than the cut-off in urine. In consultation with our service users, we agreed a cut-off for buprenorphine and norbuprenorphine of 15 μg/L.
Appropriate, and as already described, necessary, chromatographic separation was achieved. The chromatography used would also be amenable to the introduction of extra drugs in the panel in the future if a need were to develop.
The assay was successfully validated. By preparing all samples with solid phase extraction prior to LC-MS/MS analysis, matrix effects appear to have been eliminated. If we had chosen a less time-consuming, simple, protein precipitation method of sample preparation, the assay could have incurred problems due to matrix effects. However, urine is a variable matrix and it is unfeasible to test every urine sample individually for possible matrix effects. Therefore, when we process our results we are always vigilant for any unexplained suppression or enhancement of the internal standard concentrations.
The assay has acceptable precision around each drug's cut-off concentration. With the exception of 6-MAM, each drug has a lower limit of quantification below the cut-off concentration and linearity has been shown well above the cut-off concentration. We are therefore confident in reporting each drug as ‘detected’ or ‘not-detected’ when compared with the cut-off concentration. The use of a single-point calibration for each drug is sufficient for our needs and has a reduced cost compared with an unnecessary multipoint calibration.
Comparison of the new LC-MS/MS assay with our old combination of techniques showed a high level of agreement for all drugs whether positive or negative. In samples where the final result from the old methods did not agree with the final result from the LC-MS/MS method, values tended to be very close to the cut-off concentration. This discrepancy is inevitable when comparing semi-quantitative techniques.
The new LC-MS/MS assay has been in routine use in our laboratory for several months and has efficiently replaced the many stages of our previous analytical approach. The sample volume requirement, turnaround time and cost of analysis have all decreased. We can now usefully report acetyl codeine, buprenorphine and norbuprenorphine on all samples when present which we could not have done previously.
As the new LC-MS/MS panel includes measurement of morphine, 6-MAM, acetyl codeine, codeine and norcodeine, we are able to provide a comprehensive interpretation of a client's opiate intake. The metabolism of opiates is complex and as there is inter-conversion it can be difficult to determine exactly what a client has taken (Figure 4). Clients are often prescribed diamorphine or morphine sulphate. Our service users asked if we were able to identify clients that were ‘topping up’ their prescribed opiates with illicit heroin. This cannot be done using morphine and 6-MAM alone as both of these can be present due to prescribed medications. Acetyl codeine is very useful as a unique marker for illicit heroin but has a short half-life of 237 min. 14 By also measuring codeine and norcodeine on all samples, we can confidently determine whether a client has taken illicit heroin as a source of morphine. However, interpretation is still difficult if codeine has been used in addition to diamorphine or illicit heroin.
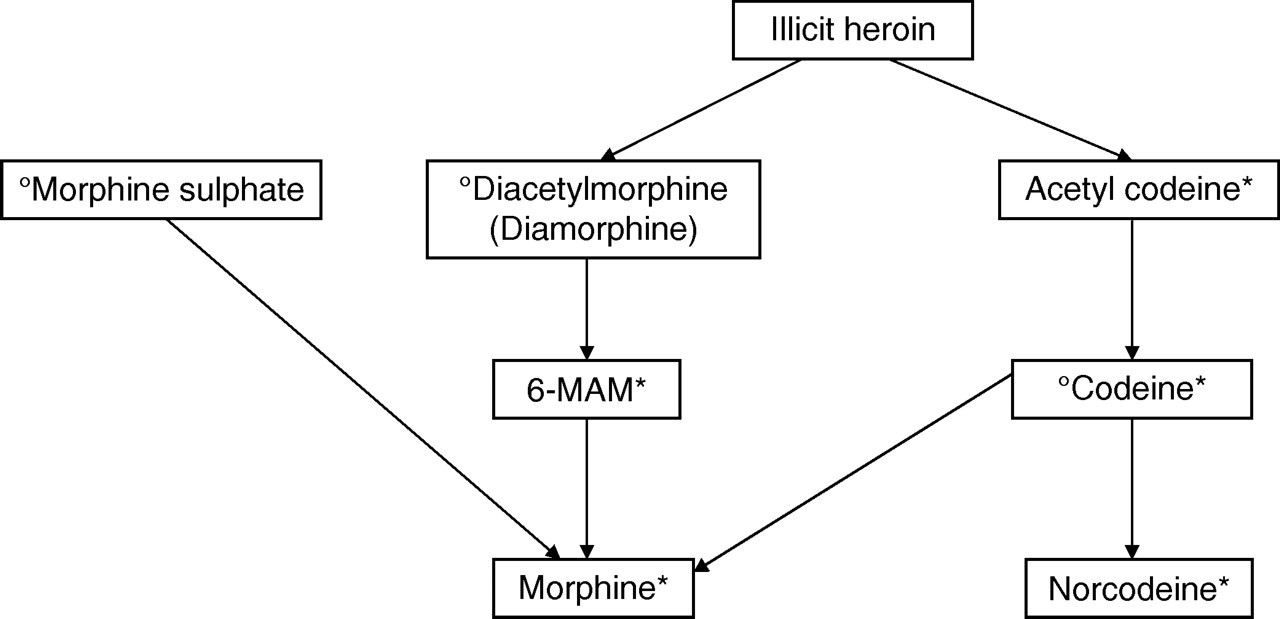
Opiate metabolism. °Denotes an opiate that may be prescribed and *denotes a metabolite that may be present in urine
Our assay is unique as it has been specifically designed to meet the needs of our service users. Similar LC-MS/MS assays have been reported, 6,15–18 but none of these would have been suitable to meet the requirements of our users. As an example, we chose not to include the cannabis metabolite, 11-nor-9-carboxy-delta-9-tetrahydrocannabinol in our panel of measured drugs as its presence in a client's urine is of no clinical interest to the community drug teams we report to.
We are continuing liaison with our local community and inpatient drug teams to maintain the contact that has been made. We organize regular meetings to ensure that our users are confident in the interpretation of the reports we produce and how to get the most amount of information from the results. This two-way contact also means laboratory staff can acquire valuable information from the drug team workers.
In conclusion, we have developed and validated an LC-MS/MS assay for 13 drugs of abuse in urine. By involving our service users, we have designed a fit-for-purpose assay that can meet their specific needs and ensure that we are providing a relevant and comprehensive service.
DECLARATIONS