
Abstract
Asymmetric dimethylarginine (ADMA) is an endogenous competitive inhibitor of nitric oxide synthase and an important cause of endothelial dysfunction. Its increased plasma concentration is associated with a variety of traditional cardiovascular risk factors, and may mediate their effects on the vascular endothelium. ADMA is also an independent predictor of cardiovascular events and mortality, and predicts outcomes in critically ill patients in the intensive care unit. This work has provided insights into the role of ADMA as an endogenous regulator of nitric oxide synthesis. At present there is no specific therapy to modify ADMA concentration, but increasing interest and work on protein arginine methyltransferases and dimethylarginine dimethylaminohydrolase, which synthesize and metabolize ADMA, respectively, might provide novel therapeutic targets.
Introduction
The endothelium plays a crucial role in the regulation of vascular tone and structure and its dysfunction is an integral part of the processes leading to atherosclerosis. The mechanisms behind this dysfunction are, therefore, of major interest to researchers. The discovery of asymmetric dimethylarginine (ADMA) and demonstration of in vivo and in vitro effects on nitric oxide synthesis has, therefore, led to a large body of work attempting to discover its role in a wide range of different clinical conditions which share endothelial dysfunction as a common feature. This research has established ADMA as a marker of cardiovascular disease, and elevated its profile as a possible mediator. It has also led to speculation that ADMA is an inherent regulator of nitric oxide synthesis. This review will provide an overview of the important clinical work which has sought to elucidate these purported roles.
Synthesis and clearance of methylarginines
Synthesis
Arginine methylation is one of many post-translational changes that contributes to the functionality of different proteins, and is a ubiquitous process. The guanidino nitrogen atoms of arginine residues within proteins are methylated by a group of enzymes called protein arginine methyltransferases (PRMTs), yielding mono and di-methylarginines. 1–3 Free arginine does not undergo methylation. Type 1 PRMTs have histone and non-histone nuclear proteins as substrates, but no activity to myelin basic protein; they are found mainly in endothelial and smooth muscle cells. 3,4 Type 2 PRMTs specifically target myelin basic protein. 3,4 ADMA is the major product of type 1 PRMTs, while type 2 PRMTs form symmetric dimethylarginine (SDMA). 4 Both are capable of producing monomethylarginine (MMA). 4 The structures of arginine and its methylated derivatives are shown in Figure 1. Protein arginine methylation has numerous physiological roles that centre around gene transcription. 4 Methylarginines appear in the cytosol on degradation of proteins containing them, and are thus a product of normal protein turnover within cells. The activity of type 1 PRMTs, and hence the rate of (asymmetric) arginine methylation, can be increased by factors such as oxidized low-density lipoprotein (LDL) 5 and increased shear stress. 6
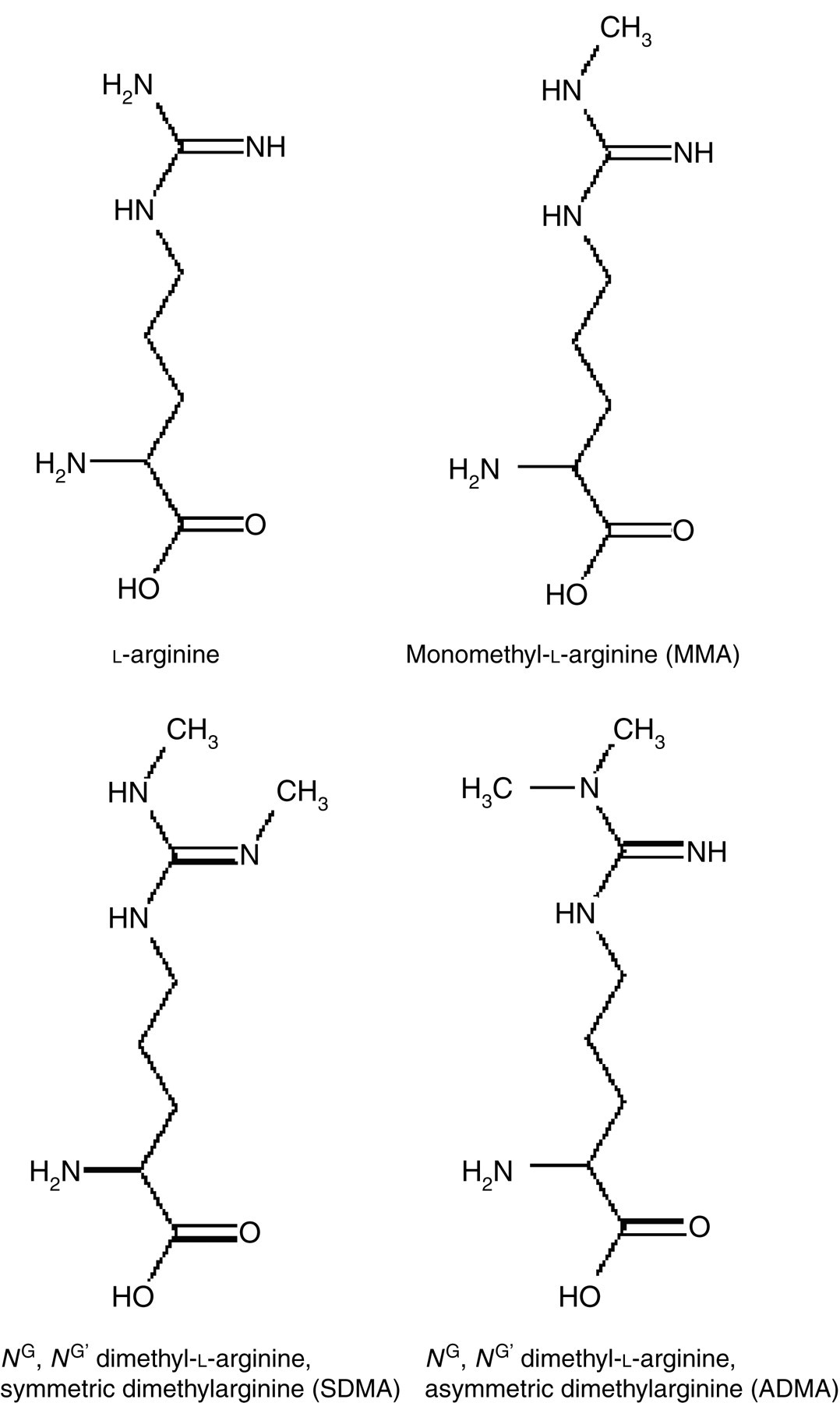
Chemical structures of arginine and its methylated derivatives
Clearance: ADMA, but not SDMA, is metabolized by dimethylarginine dimethylaminohydrolase
Humans generate approximately 300 μmol of ADMA per day, a small amount of which (∼10%) is excreted unchanged into the urine. 7,8 This was previously thought to be the main route of excretion, although study of the fates of dimethylarginines in the rabbit suggested a metabolic pathway for ADMA. 9 It is now clear that the majority (90%) is metabolized by the enzyme dimethylarginine dimethylaminohydrolase (DDAH) to yield citrulline and dimethyl-amine. 7,10,11 MMA is also metabolized by DDAH, but, in contrast, SDMA is entirely excreted in the urine, with no metabolic pathway thus far identified for its disposal. 9,10 The importance of DDAH was established by demonstrating gradual vasoconstriction in vascular rings exposed to a pharmacological inhibitor of the enzyme, an experiment suggesting that ADMA is produced continuously, with DDAH acting to prevent its accumulation. 12
Two distinct isoforms of DDAH have been identified, and have different tissue distributions, 11 a fact suggesting the importance of ADMA regulation (reviewed comprehensively by Palm et al. 11 ). DDAH-1 is found mainly in tissues expressing neuronal nitric oxide synthase (nNOS), and in tissues such as liver, kidney cortex and lung, that is those which contribute the most quantitatively to ADMA metabolism, and, thus, circulating ADMA concentration. 11,13 The kidney takes up large amounts of ADMA from the circulation, with fractional excretion of 36% in the rat kidney 14 and a net renal extraction of 16% in the human kidney, which cannot be accounted for by glomerular vfiltration alone. 15 In rat liver, the fractional excretion is around 30%, 14 and is similarly important in the human liver. 16 Indeed, in the setting of critical illness, liver dysfunction is the strongest determinant of ADMA concentration. 17
DDAH-2 is found mainly in those tissues expressing endothelial NOS (eNOS) and inducible NOS (iNOS) isoforms, and predominates in the endothelium and smooth muscle cells of the cardiovascular system. 11,13 DDAH-2 is also highly expressed in the kidney, being associated with sites of eNOS. 11 Such a distinctive distribution implies specific functions for DDAH isoforms. 11 In broad terms, DDAH-1 appears to be primarily involved in regulation of plasma ADMA concentration, and DDAH-2 with ‘local’ ADMA concentration and, thus, regulation of nitric oxide (NO)-mediated responses in the vascular endothelium. The work of Wang et al. 13 illustrates this: silencing of the mRNA for DDAH-1 significantly increased plasma ADMA concentration, but had very little effect on blood vessel NO responses. Silencing of DDAH-2, however, had no effect on plasma ADMA concentration, but significantly impaired NO-mediated responses in blood vessels. 13
The Michaelis constant (K m) of DDAH for ADMA is approximately 180 μmol/L, 10 a concentration many times that found in plasma and cells. This means that DDAH is likely to be operating within the linear part of the substrate–velocity curve; hence, changes in ADMA concentration in either direction are likely to lead to compensatory changes in DDAH activity and regulation of ADMA concentration within fairly narrow limits. The importance of DDAH is further emphasized by studies in transgenic mice over-expressing DDAH-1, which demonstrated a reduction in plasma ADMA by up to 50%, associated with increased plasma and urinary nitrates and a 15 mmHg reduction in systolic blood pressure. 18 DDAH-1 over-expression also protected against the effects of exogenous ADMA in the cerebral circulation. 19 Excess NO is also potentially harmful, particularly in acute inflammation, as the maximal velocity (V max) of iNOS is approximately 100 times that of eNOS, and can outstrip the supply of arginine, with the production of the harmful peryoxynitrite ion as a result. 20 From the foregoing discussion one would expect the biological variation of ADMA to be low; we have recently confirmed this, documenting the intraindividual variation to be 7.8%. 21
DDAH activity is attenuated by a number of oxidative stresses
The activity of DDAH is dependent on a cysteine residue at its active site. 22 This cysteine is susceptible to oxidation and nitrosylation; as a result, the activity of the enzyme can be reduced by various oxidative stresses. 23–27 Ito et al. 24 demonstrated that oxidized LDL and tumour necrosis factor-α had this effect, associated with increased ADMA concentration. 23 Similar effects have been observed with hyperglycaemia, hyperhomocystinaemia 25 and cigarette smoke extract. 26 Furthermore, through uncoupling of eNOS, increased production of superoxide could further impair DDAH, leading to a perpetuating increase in ADMA concentration. 27 The susceptibility of DDAH to oxidative stress might, therefore, provide a common pathway through which different risk factors affect the vascular endothelium. The interactions between oxidative stress, DDAH, ADMA and NOS are illustrated in Figure 2.
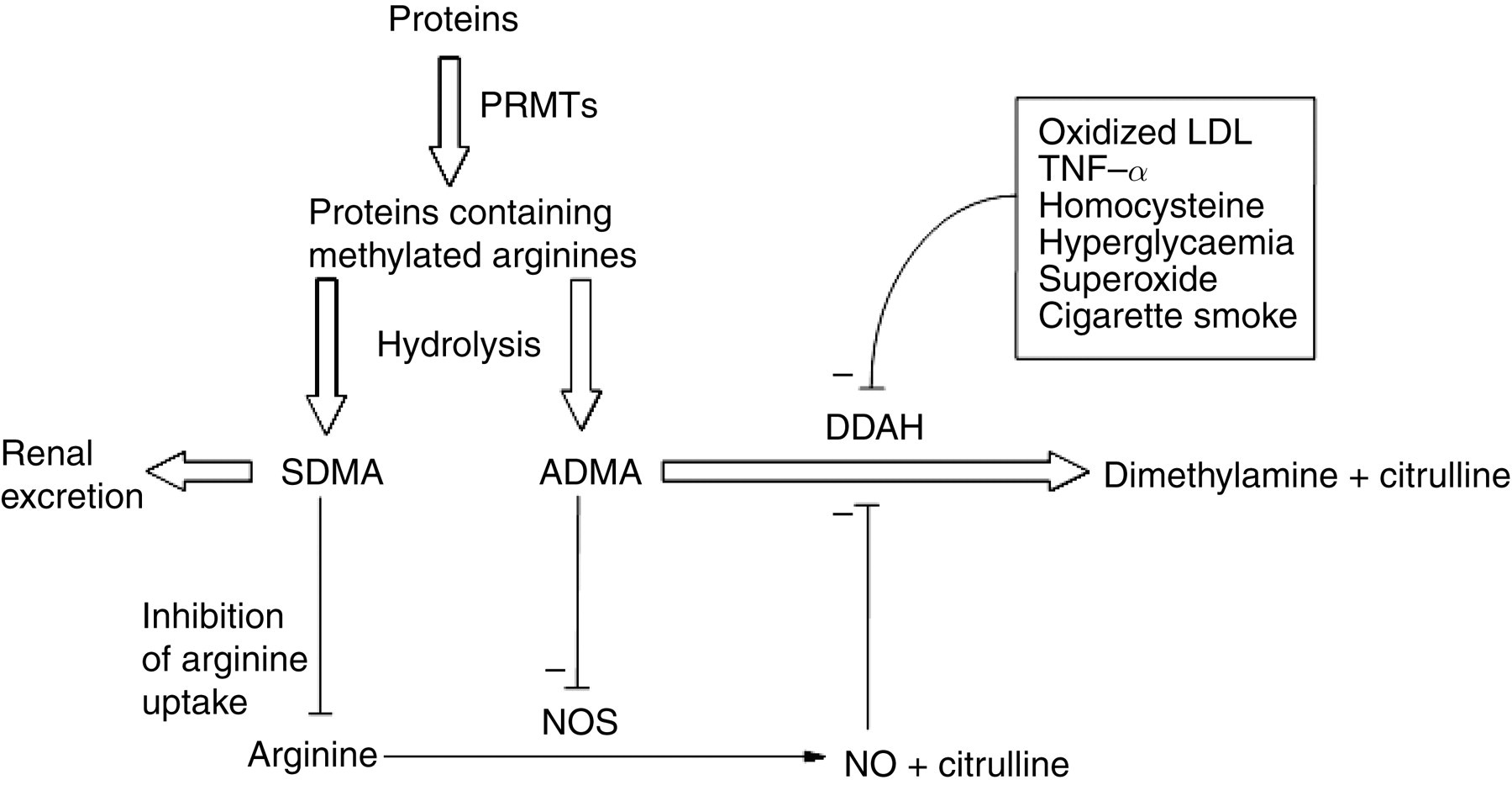
Interactions between oxidative stress, dimethylarginine dimethylaminohydrolase (DDAH), asymmetric dimethylarginine (ADMA) and nitric oxide synthase (NOS). SDMA, symmetric dimethylarginine; PRMTs, protein arginine methyltransferases; TNF-α, tumour necrosis factor-α
Pathophysiological role of ADMA
Although ADMA had been identified in biological samples decades before, 1 it was Vallance et al. in 1992 who first demonstrated a potential pathophysiological role for the molecule. They described the accumulation of dimethylarginines in patients with end-stage renal disease, speculating that increased ADMA concentration was responsible for the hypertension and immune dysfunction associated with chronic renal failure; a possible explanation for the excess in cardiovascular mortality seen in patients with kidney disease was thus advanced. 8 Since then a large volume of work has been undertaken elucidating the effects of ADMA and its role in a number of pathological states.
ADMA, but not SDMA, inhibits NOS and causes endothelial dysfunction
NO is synthesized from arginine by NOSs and is the major endothelium-derived relaxing factor, playing a critical role in the maintenance of vascular tone. 28 It has a number of other functions, including inhibition of processes involved in atherosclerosis, namely smooth muscle cell proliferation, platelet aggregation and adhesion, and monocyte adhesion; 20 it can thus be regarded as an antiatherogenic molecule. ADMA and MMA are competitive inhibitors of all NOS isoforms. 5,8,28–30 Attention has focused predominantly on ADMA as its plasma concentration is many times greater than that of MMA.
Vallance et al. 8 showed that intra-arterial infusion of ADMA into healthy volunteers caused a dose-dependent fall in forearm blood flow, an effect that could be partially offset by simultaneous infusion of arginine. In the same study, ADMA injections into guinea pigs caused an approximately 15% increase in systolic blood pressure for an ADMA concentration nine-fold higher than baseline (9.8 μmol/L). 8 In healthy males, intravenous infusion of ADMA to generate concentrations up to three times normal raised systemic vascular resistance by approximately 24% and impaired the cardiac response to exercise. 7 A similar study showed a 14% decrease in cardiac output and 11% increase in systemic vascular resistance. 31 There is also evidence of adverse effects on renal perfusion and sodium excretion, both of which are reduced by ADMA administration. 32
The ADMA concentrations associated with biological action in the experimental studies mentioned are in excess of plasma concentrations seen in healthy (for example, 2.6 7 and 5.31 μmol/L 31 compared with a ‘healthy’ value of 0.4–0.6 μmol/L 33 ) and in many disease states. This might invoke some scepticism regarding the in vivo role of endogenous ADMA. However, cells can concentrate ADMA to concentrations up to 5–10 times than that in plasma; 34 moreover, ADMA and MMA have high affinity for the amino acid transporter, competing with arginine for access to NOS. 34 From this it can be concluded that while the plasma concentration is likely to reflect the intracellular concentration, it does not in itself indicate the inhibitory concentration at the site of action. However, it is the parameter most widely documented, and the large volume of literature now available appears to confirm it as representative. Nonetheless, more work on intracellular ADMA concentrations and their inhibitory potential in pathophysiological states would help to confirm the biological significance of ADMA beyond doubt.
SDMA, in contrast, has no direct effect on NOS. However, it may still be important by competing with arginine for transport across the cationic amino acid transporter, thereby reducing substrate availability for NO synthesis. 35,36 Indeed, in cultured endothelial cells, SDMA reduced NO synthesis in a dose-dependent manner, an effect that was reversed by arginine. 36
Demonstration of the biological actions of ADMA, and the effect of oxidative stresses on DDAH, has given credibility to the concept that ADMA could explain the link between cardiovascular risk factors and endothelial dysfunction. This realization has led to much work examining the relationship between ADMA and various risk factors and clinical states.
Clinical associations
ADMA is associated with cardiovascular risk factors
Hypercholesterolaemia
ADMA concentrations are increased approximately two-fold in diet-induced hypercholesterolaemia in monkeys, 37 and are associated with endothelial dysfunction. In humans, plasma ADMA concentration has been shown to be positively correlated with cholesterol concentration, and in this context evidence of endothelial dysfunction has been demonstrated. 38 It has also been correlated with the total:HDL cholesterol ratio. 39 It should be said that other large studies have failed to replicate this link. 40–42 In one of these, however, only patients with total cholesterol <4.6 mmol/L were included, 40 and in another a significant number (>50%) were taking statins. 41 It is possible, therefore, that associations were missed in these studies. In the other, LDL cholesterol >4.1 mmol/L was associated with impaired flow-mediated dilation and reduced NO synthesis; however, ADMA concentration was not correlated with LDL. 42
There is little evidence to suggest that lowering cholesterol with statins leads to similar changes in ADMA concentration: in one study, six weeks of rosuvastatin 10 mg/d was associated with an 18% reduction in ADMA and increased flow mediated dilation. 43 Other studies have been inconsistent, with one showing a reduction in ADMA concentration following six weeks of fluvastatin 80 mg/d (endothelial function was not assessed), 44 while others showed no reduction in ADMA with statin treatment, despite significant reductions in cholesterol concentrations. 45–48 However, the response to statins, in terms of endothelial function, might be influenced by the basal ADMA concentration: higher ADMA concentrations predict a lesser effect of statins on endothelial function. 49,50 This interesting observation suggests that ADMA might be able to block the so-called ‘pleiotropic’ effects of statins, which include up-regulation of eNOS. 51 Thus, an individual's responsiveness to statins might be predetermined by their ADMA concentration, and suggests a more complex role for ADMA.
Insulin resistance and diabetes mellitus
AMDA appears to play a role in insulin resistance, and it may be that decreased NO availability helps to drive insulin resistance. 52 Hyperglycaemia impairs DDAH activity in human cells, an effect reversible by addition of a superoxide-dismutase conjugate. 24 This suggests glucose-induced oxidative stress is responsible for the effect. The plasma ADMA concentration is increased in individuals with evidence of impaired glucose tolerance, 53,54 and is associated with impaired myocardial tissue perfusion. 54 In subjects undergoing coronary angiography, ADMA was found to strongly predict glycaemic category (normal, impaired and diabetes). 55 It also predicts development of glucose intolerance in women with previous gestational diabetes mellitus, with an adjusted hazard ratio (95% confidence interval) of 3.94 (1.16–13.37) for ADMA above the median during almost three years of follow-up. 56 In obese women, ADMA concentrations are higher in those individuals with associated insulin resistance, as defined by steady-state plasma glucose; 57 weight loss through caloric restriction improves insulin sensitivity and decreases plasma ADMA concentration. 57 In women with polycystic ovarian syndrome (PCOS), the degree of insulin resistance had the greatest influence on ADMA concentration. 58 Plasma ADMA concentration is increased in patients with frank type 2 diabetes mellitus, 59,60 and is significantly higher in those with albuminuria and microangiopathy. 60
As in hypercholesterolaemia, the impact of intervention on ADMA is variable. In type 2 diabetes, three months treatment with metformin decreased plasma ADMA concentration by around 30%. 61 Metformin in PCOS also reduces ADMA, independent of its effects on glycaemic control and weight. 58 Rosiglitazone, a peroxisome proliferator-activated receptor gamma (PPAR-γ) agonist, has been shown to reduce ADMA concentration in diabetic patients in one small study, with an associated improvement in endothelial function. 53 It has also been shown to improve endothelial function in patients with type 2 diabetes without an effect on ADMA concentration. 62 Again, this suggests a more complex role, although the patients in the latter study were also taking metformin, which could have confounded the results. 62 The DDAH-2 gene contains a PPAR-γ binding site in the promoter region, and provides a possible explanation for how PPAR-γ agonists influence ADMA concentration. 63 Lastly, intensive glucose control, irrespective of how it is achieved, reduces ADMA and improves vascular function, possibly through anticytokine and antiatherogenic effects. 64
Chronic kidney disease
It is now accepted that chronic kidney disease (CKD) is a risk factor for cardiovascular disease. The increase in vascular morbidity and mortality, however, cannot be explained entirely by traditional risk factors, and ADMA might represent a novel risk factor in this context. Vallance et al. 8 were the first to demonstrate an increase in ADMA in patients with end-stage renal failure (ESRF). Since then, this observation has been replicated consistently in numerous studies of patients with CKD and ESRF (reviewed by Jacobi and Tsao 65 ). In CKD, plasma ADMA concentration is increased, with a wide range of concentrations reported (0.46–4.20 μmol/L). 65 Part of this variation is down to methodological differences: ‘control’ values of 0.36–1.40 μmol/L are reported in these studies, but, on average, ADMA is increased two-fold in CKD. 65 Although ADMA does not correlate well with serum creatinine, ADMA does increase steadily across the stages of CKD. 66 Interestingly, ADMA is also significantly increased in patients with kidney disease but normal renal function, in whom there is also evidence of increased oxidative stress. 66,67 This, plus evidence from prospective studies, suggests that ADMA might play a role in progression of renal disease, its concentration strongly predicting progression to ESRF. 68,69
ADMA is also a determinant of cardiovascular disease in CKD. It is independently associated with carotid intima–media thickness (IMT) and predicts its progression. 70 It is also associated with left ventricular hypertrophy and cardiac dysfunction. 71 In CKD, ADMA predicts death independently of other variables. 68,72 As little as a 0.1 μmol/L increase in plasma ADMA is associated with a 20% increase in a combined endpoint of ESRF and death on multivariate analysis. 68 In haemodialysis patients, ADMA is the second strongest predictor of death after age, with an ADMA concentration above the 75th centile conferring a two- to three-fold increased risk of death. 72
The reason for the increase in ADMA concentration in CKD is likely related to two major factors. Firstly, given the high net renal extraction in humans, which is around 16% and primarily related to DDAH-mediated catabolism, 15 ADMA can accumulate due to decreased metabolism, and could be regarded as a metabolic complication of CKD, much like bone disease and anaemia. Secondly, CKD is associated with increased oxidative stress, with correlations between ADMA and markers such as plasma malondialdehyde, erythrocyte superoxide dismutase and oxidized LDL. 66 As discussed above, oxidative stress is associated with reduced DDAH activity and ADMA accumulation.
Unlike ADMA, SDMA correlates highly with glomerular filtration rate (GFR) as determined by inulin clearance (r = 0.85, P < 0.0001), and appears to be a more sensitive marker of renal function than creatinine; 73 as a sensitive and non-invasive marker it might have a future role. Its indirect effects on NO production have already been alluded to, and in this context it is interesting to note that its concentration is correlated with the angiographic stenosis score in patients with mild CKD. 36 Therefore, although SDMA has no direct effect on NOS, it might still be an important measure, firstly, as a sensitive marker of GFR and, secondly, as a tool for estimating cardiovascular risk in early CKD. 36
Hypertension
In humans with essential hypertension, ADMA concentration is increased two-fold over healthy controls and is associated with reduced levels of urinary NO metabolites. 74 ADMA concentration correlates with markers of endothelial function in hypertensive patients. 75
In healthy young men, black Africans have significantly reduced NO and flow-mediated dilation compared with white Europeans, and this is independently associated with ADMA concentration. 76 Treatment guidelines for hypertension acknowledge that black African and Caribbean patients are less responsive to inhibition of the renin-angiotensin system than white patients, 77 revealing what is likely a fundamental difference in the pathophysiology of hypertension in these groups.
Several studies have demonstrated reduced ADMA concentrations with short-term angiotensin converting enzyme (ACE) inhibitor treatment; in these studies, ADMA concentration was reduced by 12–20%, and included patients with essential hypertension, 78–80 insulin resistance, 81 diabetes mellitus 82 and CKD. 83 Angiotensin-receptor blocking drugs produce similar reductions in ADMA concentration, and can improve vascular function. 78,80,84 There is some evidence that this might be related, at least in part, to inhibition of the effects of oxidative stress, such as lipid peroxidation, and so restoration of DDAH activity. 85 This may be a factor in explaining the protective effects of ACE inhibitors on the vascular endothelium.
ADMA and cardiovascular disease
ADMA concentration is associated with the presence and extent of atherosclerotic vascular disease. In a large multicentre case-controlled study, ADMA had the best discriminative power to distinguish cases of coronary heart disease (CHD) from controls, and in a multivariate model was a positive predictor with an odds ratio (95% CI) of 6.04 (2.56–14.25) for those in the highest tertile of ADMA versus the lowest (>0.72 and <0.58 μmol/L respectively). 86 In men with early stage atherosclerosis, ADMA was associated with the presence of measurable endothelial dysfunction: in this study, patients with endothelial dysfunction also had a higher prevalence of erectile dysfunction, something which could be related to reduced NO availability. 87 Moreover, in those with established CHD, it is associated with the severity of lesions at angiography. 88 ADMA concentration gradients have been demonstrated in coronary vessels, with ADMA concentrations significantly higher distal to atherosclerotic plaques than proximal to them (2.39 versus 1.52 μmol/L). 89 ADMA correlates significantly with carotid IMT, and independently predicts its progression. 70,90 A nested case-control study of healthy non-smoking men showed that ADMA concentration in the highest tertile (>0.62 μmol/L) conferred an almost four-fold increase in the incidence of acute coronary events, after adjustment for other factors. 91
Prospective studies have provided what is arguably the most important evidence establishing the role of ADMA in cardiovascular disease, repeatedly showing its predictive value, independent of both traditional and novel cardiovascular risk factors. This includes prediction of acute coronary events, cardiovascular mortality and all-cause mortality, and has been demonstrated in those with coronary artery disease, 92,41 following myocardial infarction, 93 chronic heart failure, 94 peripheral arterial disease, 95 type 1 and type 2 diabetes, 96,97 following percutaneous coronary intervention 98 and in the general population. 99–101 The major findings of these studies are summarized in Table 1, with hazard ratios adjusted for traditional risk factors. In at-risk individuals and in those with a prior history of CHD, increased ADMA concentrations confer about a two-fold increase in the risks of cardiovascular events and death. The same is true in general population studies, with 20–30% increases in coronary events 99,100 and all-cause mortality, 101 whose associations are linear. In many of them, increased risk is demonstrated at concentrations of ADMA that could be classified as ‘normal’ by many assay methodologies for ADMA, and highlights the issue of inaccurate methodology, as well as the limitation of traditionally defined population reference intervals. Both of these are discussed later in this article.
Evidence from prospective studies showing ADMA as a prognostic marker
CHD, coronary heart disease; MI, myocardial infarction; CHF, congestive heart failure; PAD, peripheral arterial disease;
CV, cardiovascular; PCI, percutaneous coronary intervention;
DM, diabetes mellitus; SD, standard deviation;
CI, confidence interval; ADMA, asymmetric dimethylarginine
ADMA concentrations are increased in patients with acute coronary syndromes (ACS) compared with those with stable angina, and decrease rapidly with medical treatment. 102 Moreover, the failure of ADMA to decrease six weeks following percutaneous intervention for ACS strongly predicts a recurrent event during one year of follow-up. 103 As well as a prognostic indicator, these studies suggest ADMA to have an important role in the development of cardiovascular disease, both acutely and chronically.
ADMA in critical illness
ADMA concentration independently predicts mortality in critically ill patients in the intensive care unit (ICU). In a prospective study of 52 patients, ADMA was the strongest predictor of outcome, with a 17-fold increase in mortality for patients with ADMA concentrations in the highest quartile; 17 in this study, markers of hepatic dysfunction were the strongest determinants of ADMA concentration. In another similar study, ADMA concentration was correlated with organ failure, shock and inflammatory markers. 104 In rats, the liver takes up large amounts of ADMA, amounting to several hundred times that circulating in plasma; 14 in contrast SDMA is not taken up, suggesting that metabolism by DDAH is the mechanism for hepatic ADMA clearance. 14 In rats with lipopolysaccharide-induced endotoxaemia, a reduced ADMA concentration is observed, and is associated with increased fractional excretion by the liver (41.0% versus 27.7%), suggesting up-regulation of metabolic clearance. 105 Hepatic function, therefore, appears to play a key role in ADMA regulation especially during acute inflammation.
It has been proposed that ADMA contributes to the development of multiple organ failure in critical illness. 106 It seems likely that a combination of ADMA liberation from increased protein turnover and accumulation from renal and hepatic failure act together to increase its concentration. The effect of oxidative stress on DDAH activity has already been described, and this could be a major reason for ADMA accumulation. Through inhibition of eNOS in particular, constitutive NO production is decreased, causing disturbance of organ blood flow and decreased cardiac output, as well as other effects including capillary leakage and thrombocyte aggregation. 106
Intensive insulin therapy to maintain normoglycaemia has been shown to improve ICU outcomes. 107 It has been proposed that modulation of ADMA concentration by insulin could account for this beneficial effect. 108 Intensive insulin treatment was associated with ADMA concentrations that were lower on day 2 than those treated conventionally; moreover at the end of the ICU period, intensively treated patients had lower ADMA concentrations, and there was a correlation between total insulin dose and ADMA concentration on the last day. 108 These findings could be explained by preservation of DDAH activity, since hyperglycaemia inhibits DDAH. 24 Other explanations include reduced protein catabolism and increased uptake of ADMA into cells through up-regulation of the cationic amino acid transporter. 109
It has already been stated that excessive NO production can be harmful, and has been implicated in the development of septic shock. 110 Because of this, inhibition of iNOS with MMA was advanced as a possible way of reducing mortality. However, a clinical trial actually demonstrated increased mortality. 111 It might be that non-specific inhibition of NOS isoforms – particularly eNOS – was responsible for the excess deaths. The different tissue distributions of DDAH isoforms, and their co-localization with NOS isoforms, might point to differential inhibition of NOS isoforms and hence an important regulatory role for ADMA in the control of NO production in inflammation. Further evidence for this possible role is provided by the attenuation of DDAH activity by NO itself. 112 Furthermore, endothelial cell and macrophage uptake of NOS inhibitors (ADMA and MMA) is increased in response to an inflammatory stimulus. 113 These latter two observations theoretically permit intracellular ADMA (and MMA) concentrations to increase above what would otherwise be encountered, thus forming the basis of an elegant negative feedback system controlling NO production (Figure 2). Further work in models of inflammation is required to further elucidate the role of ADMA.
Measurement of ADMA
The measurement of ADMA in plasma is rendered difficult by its low (submicromolar) concentration, and in achieving chromatographic separation from its structural isomer SDMA. High-performance liquid chromatography (HPLC) is the most widely used method, 114–120 the majority employing reverse-phase chromatography with fluorescence detection; ortho-phthaldialdehyde is generally employed as a derivatizing agent since ADMA itself is not fluorescent. Complete separation of ADMA and SDMA can be achieved under isocratic conditions, 114,115,118,119 although it is sometimes necessary to use gradient elution. 116,117,120 Homoarginine and MMA are the most commonly used internal standards. However, both are inherent in human plasma, homoarginine being present in concentrations up to 5 μmol/L. 114,115 Moreover, MMA concentration increases in renal failure, and its use precludes quantification of endogenous MMA, something that may be of importance in future work on intracellular concentrations. 34 We have recently validated an HPLC method using the novel internal standard monoethylarginine which is not inherent in human plasma, and so overcomes these problems. 114 The major drawbacks of HPLC are relatively long analysis times and the necessity for sample clean-up, which involves solid-phase extraction. However, they offer the major advantage of permitting reliable quantification of ADMA, SDMA and arginine on a single sample. As can be seen from Table 2, most HPLC methods offer acceptable precision, and, in the main, population reference values derived using them are fairly similar.
Methods for quantification of asymmetric dimethylarginine (ADMA) in human plasma
OPA, ortho-phthaldialdehyde; AccQFluor™, 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate; NDA, naphthalene-2,3-dicarboxaldehyde; PFP, pentafluoropropionyl; SD, standard deviation; CV, coefficient of variation; ELISA, enzyme-linked immunosorbent assay; LC-MS/MS, liquid chromatography-tandem mass spectrometry; RP HPLC, reverse phase high-performance liquid chromatography
*Reference values presented stratified into four groups according to age. Mean and SD of the whole group not given
†SD not given. Range quoted between 10th and 90th centiles
Mass spectrometry (MS) methods are increasingly described. 121–127 Because of its specificity, the use of MS allows laborious sample clean-up techniques to be omitted, with only a simple protein precipitation step remaining necessary. Separation by gas or liquid chromatography or derivatization are necessary steps because the fragmentation patterns of ADMA and SDMA on tandem MS are very similar. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) is most commonly used, but gas chromatography has also been employed. Most MS methods report mean population reference values within the range 0.39–0.66 μmol/L, as can be seen in Table 2. Apart from increased specificity, these methods permit simpler sample preparation and reduced analysis times. However, not all achieve desirable precision, and, because of the equipment and expertise required, might not be universally available.
An enzyme-linked immunosorbent assay (ELISA) assay has been developed and validated, 128 and has been used in large trials. The major advantage of ELISA is the high throughput of samples that can be achieved. However, results obtained by ELISA are significantly higher than those from robust chromatographic methods, as has been shown in comparisons with HPLC 129,130 and LC-MS. 131 On average, ELISA yields concentrations approximately twice as high as those from chromatographic methods, suggesting matrix effects or calibration problems. 129,131 One group performing a comparison, however, found no correlation between the methods. 130 Another major drawback is the relatively high imprecision of the method, which was 10% in the described assay validation, 128 but can be higher when used by others. 130 Indeed, in two large prospective trials using ELISA, the differences between controls and cases were relatively small (≤0.1 μmol/L), 41,86 and not significantly different from a population reference value derived using this method. 132 One final drawback is the inability to measure SDMA and arginine, which both HPLC and MS methods allow.
In summary, ADMA methods produce significantly different results, as can be seen from the variation in described reference values in various clinical studies, illustrated in the review by Jacobi and Tsao, 65 and in Table 1. The low interindividual biological variation of ADMA (9.6%) 21 strongly suggests that these differences are largely due to method specific factors. A recent comparative exercise, the only one done to date, illustrates and highlights this problem. 33 The effect of it has been to hinder the collation of the large amount of ADMA research. Vallance and Leiper 133 cite the lack of standardization as the major reason why a meta-analysis of research in patients with renal disease cannot be performed, something that would help to better quantify the risk associated with ADMA. This variation is a barrier to the clinical application of ADMA, as concentrations in various disease states and in health have not been defined clearly.
Biological variation of ADMA and analytical performance goals
The concentrations of both ADMA and SDMA are tightly controlled in health, with intraindividual variations (CVI) of 7.4% and 5.8%, respectively. 21 Based on a desirable imprecision of no more than 0.5 times CVI, 134 it is clear that many assays fail to meet this goal. The effect of increasing imprecision has been highlighted by Teerlink: 135 increasing imprecision was simulated by adding an error term to data obtained from a real experiment. The results demonstrated a rapid falling off of the number of experiments reaching statistical significance with coefficients of variation (CV) in the range 5–10%, which some people might view as being acceptable. This is especially true for studies containing a relatively small number of subjects. The author rightly concludes that the effect of imprecision is to miss potentially important findings in clinical studies, and to undermine associations between ADMA and other parameters. 135
Population reference intervals
There have been relatively few population studies encompassing a reasonable number of well-defined ‘healthy’ individuals. We recently reported a 95% reference interval of 0.29–0.63 (mean 0.45) μmol/L, 21 which is broadly similar to that derived from 2311 individuals by Teerlink, who reported 0.39–0.63 (mean 0.50) μmol/L using a similar HPLC method. 136 Schulze reported a 95% reference interval of 0.36–1.17 (mean 0.69) μmol/L in 500 individuals using ELISA. 132 The higher mean value is consistent with what has already been said about the comparison between ELISA and chromatography. The significantly wider range most likely reflects the greater imprecision of the assay. Other studies of greater than 100 individuals have reported results that equate to 95% ranges of 0.30–0.82 (mean 0.51) μmol/L137 and 0.32–0.62 (mean 0.50) μmol/L, 138 both using HPLC. Overall, a mean concentration of around 0.50 μmol/L seems to be close to the reference population value.
The data of Meinitzer et al. 115 Schulze et al. 132 and Teerlink 136 indicate that ADMA is positively correlated with age; furthermore, they have shown increased concentrations in postmenopausal women compared with men of a similar age. However the increase, though statistically significant, is biologically small, and it has been suggested that the generation of age- and gender-specific reference intervals would be of little, if any, benefit. 132 Meinitzer et al. 115 has described reference intervals stratified according to age, and the differences are small, with means ranging from 0.58 to 0.64 μmol/L.
We have previously discussed the usefulness of population reference intervals for ADMA. 21 Given its high individuality (index of individuality 0.81), population reference intervals are unlikely to be useful in denoting ‘abnormality’ from a single result; 21,139 this is analogous to serum creatinine. Single measurements are, therefore, likely to be difficult to interpret in the context of cardiovascular risk assessment, for example, and almost certainly precludes the use of ADMA as a standalone composite risk marker. Reference to the ADMA concentrations in the studies summarized in Table 1 emphasizes this point.
Conclusions
The discovery of NO was a major breakthrough in understanding the biology of the vascular endothelium. The discovery of an endogenous molecule that can influence its production, therefore, is also of major importance. There is now abundant evidence establishing ADMA as a marker, if not mediator, of cardiovascular disease. However, it has not yet established itself as a clinically useful measurement, despite many prospective studies demonstrating its strong predictive power for cardiovascular events and mortality, independent of traditional risk factors. There are two reasons for this. Firstly, there has been considerable uncertainty over what constitutes a ‘normal’ concentration, and the degree of risk associated with an increase in concentration is, therefore, difficult to quantify; furthermore clinically significant increases can occur within a range that could be considered ‘normal’. Secondly, and more importantly, there is no therapy currently available to specifically modulate ADMA concentration. ADMA concentrations can be lowered by well-established treatments such as metformin, rosiglitazone, insulin, statins and ACE inhibitors, and might provide some insight into the way these drugs exert their beneficial effects. However, it will require the development of a specific agent to modify ADMA concentration, and proof that this confers benefit, to elevate the importance of ADMA above other markers such as high sensitivity C-reactive protein and homocysteine. Nonetheless, further evaluation of the role of ADMA as a means of improving cardiovascular risk assessment would be interesting, particularly given its prognostic significance.
Future research is likely to focus on the enzymes DDAH and PRMT and the genetics that determine an individual's ADMA concentration. This will increase understanding of the role of ADMA and potentially provide therapeutic targets. More work on the role of ADMA in critical illness, in particular, might yield observations fundamental to the understanding of NO and its control in that setting, and, again, provide targets for treatment in a group of patients with a high mortality.
DECLARATIONS