
Abstract
Background
Up to 2007 there was no formal external quality assurance programme for plasma free metanephrines. A pilot programme was conceived by the AACB (Australian Association of Clinical Biochemists) Working Party on biogenic amines. With support from the AACB and Royal College of Pathologists of Australasia Quality Assurance programmes, a pilot study was developed. Data from this study are presented for the first time.
Methods
Twelve lyophilized plasma samples were distributed to 15 centres. Samples were spiked with metanephrine (metadrenaline), normetanephrine (normetadrenaline) and 3-methoxytyramine, all derived from human urine. Concentrations were arranged in a linear relationship. The analytes were present at six levels and samples were duplicated.
Results
High-pressure liquid chromatography and tandem mass spectrometry methods showed acceptable precision but in general enzyme immunoassay displayed a higher degree of imprecision as well as a negative bias.
Conclusions
Differences in calibration and matrix effects are likely to have been responsible for the discrepancy between chromatographic and immunoassay methods. These differences need to be further examined although efforts at standardization between different methods have been hampered by the lack of a universal calibrator for plasma metanephrines. Meanwhile, a laboratory's performance characteristics can be monitored and enhanced by participation in suitable external quality assurance programmes.
Introduction
Laboratory measurements of catecholamines and their metabolites in urine have long been the diagnostic test of choice for biochemical diagnosis of phaeochromocytoma. The catecholamines – noradrenaline and adrenaline – are metabolized to the metanephrines – normetanephrine and metanephrine and 4-hydroxy-3-methoxy mandelic acid (HMMA). A 24-h urine sample is necessary to allow for circadian variations in catecholamine secretion. For many years there has been a preference for estimations of catecholamines and HMMA as frontline tests for evidence of the tumour. There is now, however, a substantial and growing body of evidence that metanephrines display superior sensitivity over catecholamines and HMMA for this purpose. 1–4 While there is continuing debate regarding the relative merits of urinary versus plasma metanephrines, there is emerging consensus that either plasma free or urinary fractionated metanephrines should be adopted as the frontline test for the tumour. 2–6 This and many other issues relating to aspects of biochemical screening for phaeochromocytoma have been addressed in a recently compiled comprehensive review. 5
Plasma metanephrines
A practical method for the analysis of plasma free metanephrines was described by Lenders et al. 7 Its usefulness in the diagnosis of phaeochromocytoma was demonstrated in a study designed to identify the biochemical test or combination of tests that provided the best method for this purpose. 1 Using ROC (receiver operating characteristic) curves it was shown that sensitivity and specificity values at different upper reference limits were highest for plasma metanephrines.
Free metanephrines in normal plasma are present only in sub-nanomolar amounts and accurate measurements call for sensitive instrumentation and careful analytical techniques. As a consequence, the numbers of centres measuring plasma metanephrines are still limited. Current methods include high pressure liquid chromatography (HPLC) with coulometric detection (CD), 7 immunoassay 8,9 and liquid chromatography-tandem mass spectroscopy (LC-MS/MS). 10,11
Quality assurance
External quality assurance (EQA) schemes for urinary biogenic amines have been provided for many years by agencies such the National External Quality Assurance Scheme (UK), Royal College of Pathologists of Australasia (RCPA) Chemical Pathology Programs and the American Association of Clinical Chemists. However, there have been no such schemes involving plasma metanephrines until a recent proficiency survey facilitated by the EQA provider ProBioQual (France). 12 This trial involved the distribution of four lyophilized samples to 10 centres. Two of the samples were prepared in a citrated plasma matrix with the other two prepared in a serum matrix. The study revealed important method-related differences in results and a wide distribution of results especially for metanephrine. Also evident was the difficulty involved in evaluation of the results in the absence of a commercially available calibrator.
Despite the advantages of the plasma metanephrine test, its availability is still limited. It is possible that the lack of adequate EQA programmes that include the test may have contributed to this situation. The Australian Association of Clinical Biochemists (AACB) Working Party on Biogenic amines sought to conduct an extended pilot quality assurance programme for plasma free metanephrines. It was planned to serve as a precursor to a comprehensive EQA programme that would be conducted annually on an ongoing basis. In addition to free normetanephrine and metanephrine, it included the dopamine metabolite 3-methoxytyramine which has been proposed as a diagnostic marker for rare dopamine secreting paragangliomas. 13
Method
Support for the trial was sought and obtained from the Scientific and Regulatory Affairs committee of the AACB and from RCPA Chemical Pathology Quality Assurance Programs. The latter was to assume responsibility for administration and data management. Expressions of interest were canvassed from centres known to be involved in estimations of plasma metanephrines. A total of 15 laboratories sought enrolment in the pilot programme. They included key centres in Australia, Germany, New Zealand, Switzerland, the Netherlands, UK and US.
The samples were prepared by Australian Scientific Enterprise, an organization that has been responsible for production of material for EQA programmes including RCPA Chemical Pathology Quality Assurance Programs. Human plasma was supplemented with metanephrine, normetanephrine and 3-methoxytyramine. These substances were obtained by hydrolysis of human urine. No stabilizers or preservatives were added before lyophilization. It was assumed that only L-isomers of these compounds were present in the supplemented urine material. This presumption was based on the absence of evidence in the literature for the presence of measurable amounts of D-isomers in human urine. It is likely that the preparation of commercial calibrators for immunoassay kits have also relied on this premise. The suitability and chemical integrity of representative samples for the programme was assessed in laboratories of AACB Working Party members. The assessment was conducted before and after lyophilization using HPLC with CD.
The lyophilized material was shipped to enrolled laboratories in March 2008. Twelve samples were supplied to each centre with the requirement that two samples would be analysed every four weeks and results were submitted thereafter. Samples provided to participants spanned six linearly related concentration levels for each analyte. Duplicate samples of each concentration level were assayed on separate occasions by each laboratory. There was provision for submission of data by mail or electronic media. Reports were posted to participants usually within a few days of the monthly due date for each pair of specimens. The reporting format employed for this pilot EQA programme followed the standard format employed by other RCPA EQA programmes and is well described on the RCPA Chemical Pathology QAP website.
For the purposes of this study, precision of specific method groups was assessed as follows:
Linear regression lines were plotted using each laboratory's duplicate values for each of the six linearly related concentration levels; The standard error of the estimate (Sy.x) for the laboratory's results was calculated using this regression line; The midpoint of the laboratory's range of measured concentrations was calculated by interpolation from its highest and lowest reported concentrations; The % coefficient of variation (%CV) (intra-laboratory) was calculated by dividing Sy.x by the midpoint concentration.
The use of the standard error of the estimate calculated from each laboratory's own regression line provides the benefit of representing intra-laboratory precision regardless of any bias experienced by that laboratory.
There is currently no standard reference material or reference technique listed on the Joint Committee for Traceability in Laboratory Medicine database for the measurement of free plasma metanephrines. Therefore it was not possible to determine bias versus an absolute reference. However a visual summary of laboratory bias is presented in Figures 1 –3 showing the regression lines of all laboratories. The line of identity on these graphs represents the median of all results submitted for the analyte. For all three analytes these medians are derived from the HPLC-CD method group. Therefore these graphs broadly represent each laboratory's bias versus this method.
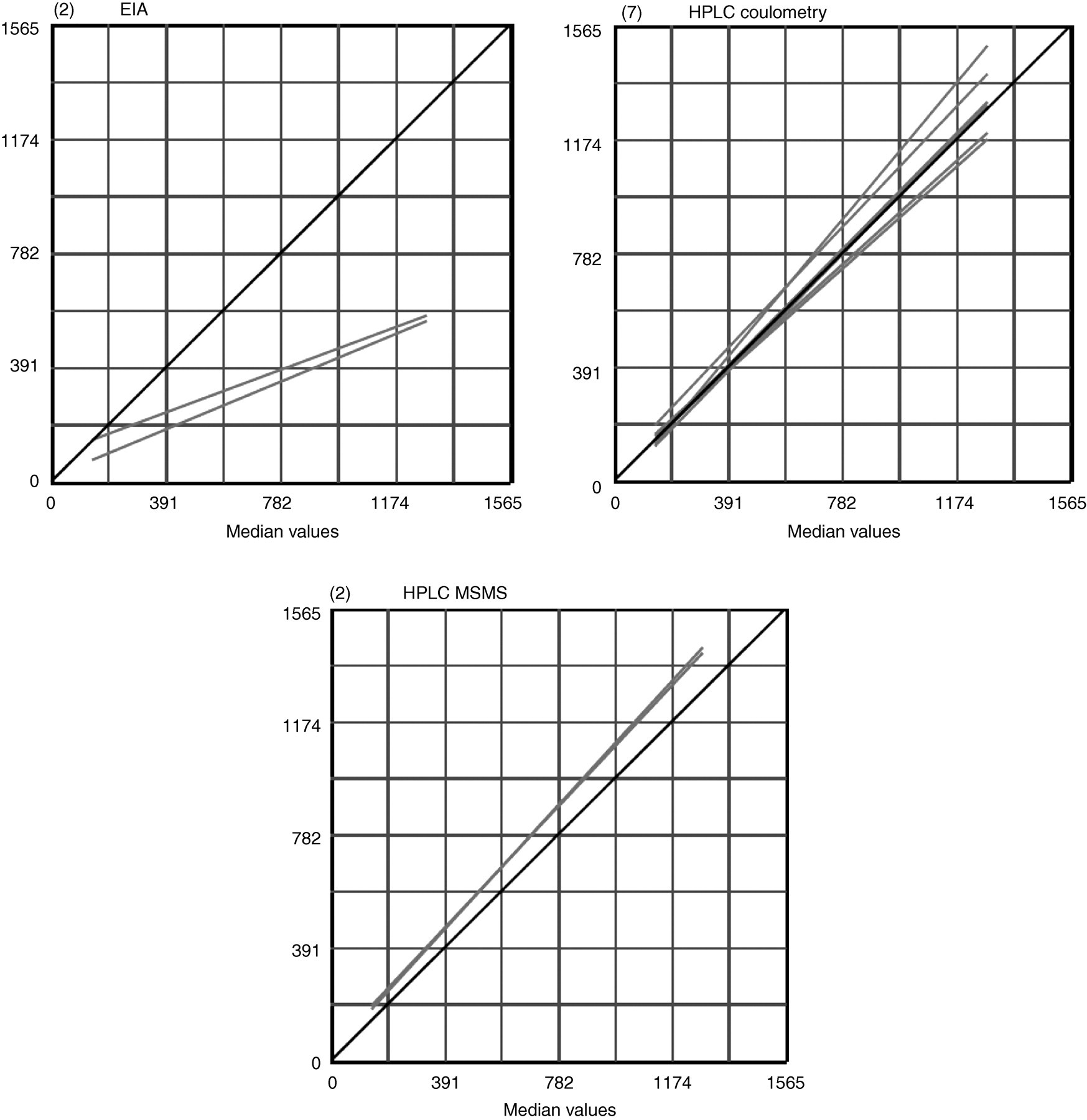
Plasma free metanephrine (nmol/L). Linear regression lines for EIA, HPLC coulometry and HPLC MSMS. The number in brackets at top left of each graph shows the number of laboratories in the method group
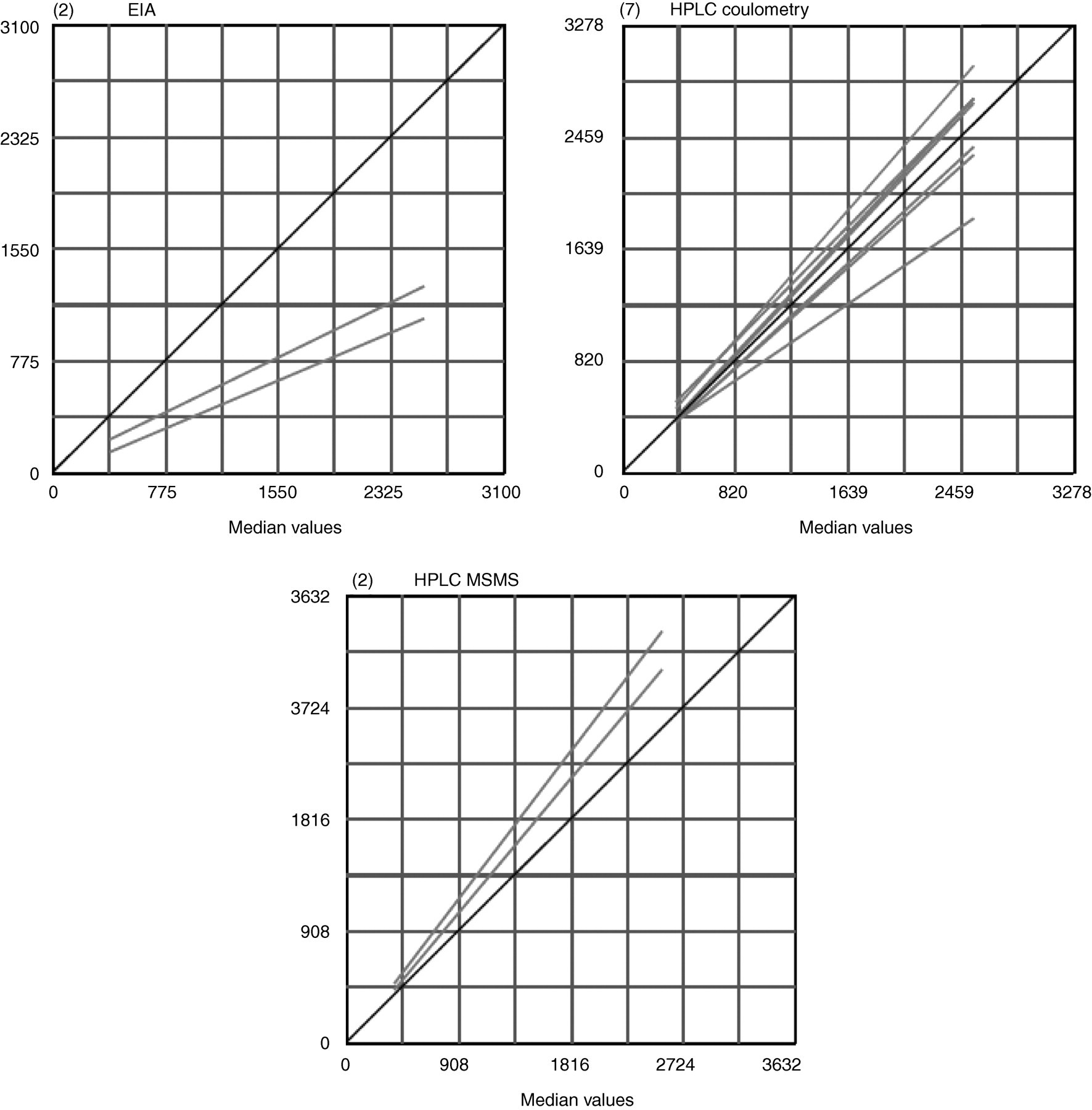
Plasma free normetanephrine (nmol/L). Linear regression lines for EIA, HPLC coulometry and HPLC MSMS. The number in brackets at top left of each graph shows the number of laboratories in the method group
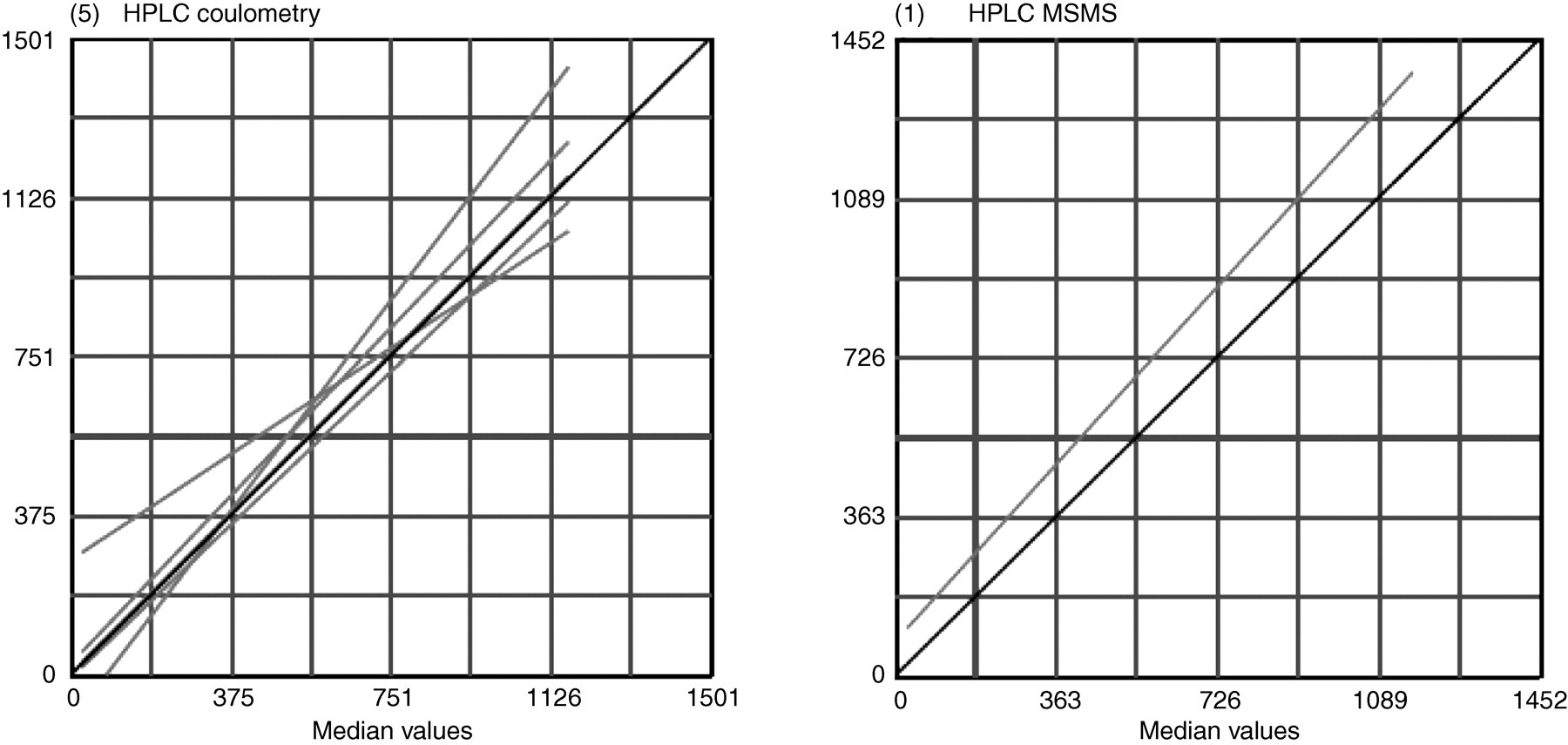
Plasma 3-methoxytyramine (nmol/L). Linear regression lines for EIA, HPLC coulometry and HPLC MSMS. The number in brackets at top left of each graph shows the number of laboratories in the method group
Bias has also been assessed for each method group by calculating the percentage difference of each group median from the overall median of all laboratories at the highest and lowest concentration levels. This is shown in Table 1. Again this represents percent bias from the overall median, where this has been derived from the HPLC-CD method group for all three analytes.
Summary of imprecision and relative bias
CV, coefficient of variation; HPLC-CD, high-pressure liquid coulometric detection chromatography
Results
Results were returned by 11 of the 15 enrolled laboratories. It was assumed that enrolled centres that did not return results were availing themselves of samples to develop or improve their assays. Of these 11 participants, seven used HPLC-CD, two used LC-MS/MS while two utilized enzyme immunoassay (EIA) techniques. 3-Methoxytyramine was reported by six centres (5 HPLC-CD and 1 LC-MS/MS).
The median of the intra-laboratory CV% for each of these groups is shown in Table 1. The median CV% for both HPLC-CD and LC-MS/MS methods show acceptable precision for metanephrine and normetanephrine. HPLC-CD also shows acceptable precision for 3-methoxytyramine. There is difficulty in assessing the precision for LC-MS/MS determination of 3-methoxytyramine as only a single intra-laboratory CV% was available. The EIA technique displays a high level of imprecision for both metanephrine and normetanephrine measurements.
The EIA group shows a marked negative bias for metanephrine and normetanephrine at both the high and the low concentration levels relative to the HPLC-CD methods (Table 1). Regression lines (Figures 1 and 2) of the two EIA laboratories also show this negative bias which is proportional to concentration for metanephrines, but appears more constant for normetanephrine.
The LC-MS/MS method shows a marked positive bias at lower metanephrine concentrations relative to HPLC-CD. In general the LC-MS/MS method gave slightly higher values at high concentrations of metanephrine and at all levels of normetanephrine. It is difficult to draw any conclusions regarding bias for 3-Methoxytyramine methods due to limited data.
Overall, there was generally good agreement between laboratories using similar methodology. Assessment of the true bias of methods cannot be made until a traceable calibrator for plasma metanephrines becomes available.
Discussion
The study showed important differences in results between chromatographic and immunoassay methods. These differences are likely to have arisen from differences in calibration procedures. However matrix effects may also have contributed to the discrepancies between the techniques. In any case these findings need to be treated with caution due to the small number of laboratories that participated in the study.
The ProBioQual proficiency survey 12 showed that an immunoassay method gave lower concentrations of normetanephrine even after a correction for the amount of L-isomer in the sample. However, levels of metanephrine were found to be relatively similar for immunoassay and chromatographic methods. Again, it is likely that calibration and matrix effects to a lesser extent may have been responsible for the disparity. The contribution of matrix effects can be clarified by direct comparisons of identical samples of normal plasma by immunoassay and HPLC or LC-MS/MS methods.
Of great concern is the validity of reference intervals quoted for immunoassay methods. The upper limits of reference intervals for normetanephrine quoted by certain immunoassays exceed those of HPLC methods and especially when samples are taken in the supine position. There is therefore potential for false-negative results with some immunoassays if they are underestimating normetanephrine. It is therefore imperative that further investigations be conducted so that these issues can be clarified.
This study also demonstrated the need for a universal calibrator for plasma metanephrines to enable the comparison of results obtained by multiple methods. For immunoassays, it is essential that such calibrators are prepared using the natural L-isomers. Meanwhile, it is important that centres performing the plasma free metanephrine test ensure participation in appropriate EQA programmes such as the current RCPA program which evolved from this pilot study. The 2009 RCPA EQA scheme appears to be the only currently available programme for plasma free metanephrines. It is ongoing. Participation in such EQA programmes can not only generate confidence in the validity of their results but may well contribute towards compliance with any regulatory requirements.
DECLARATIONS
The participation of the following institutions is also acknowledged:
Chemical Pathology Department, Pathology Queensland–Central Herston Hospitals, Herston, Queensland 4006, Australia; Biochemistry Department, Pathwest, Royal Perth Hospital, Perth WA 6847, Australia; Clinical Chemistry Department, The Prince of Wales Hospital, Randwick, NSW 2031, Australia; SA Pathology, Flinders Medical Centre, Bedford Park, SA 5042, Australia; A.R.U.P Laboratories, Salt Lake City, UT, USA; Inst Clin Chem and Lab Medicine, University Hospital Carl Gustav Carus Dr, Dresden, Germany; Chemical Endocrinology, Raboud University Nijmegen Medical Centre, Nijmegen, Netherlands; Clinical Biochemistry, Freeman Hospital, Newcastle upon Tyne, UK; Division of Clinical Pharmacology and Toxicology, Chuv/Hospital Nestle, Lausanne, Switzerland; Canterbury Health Laboratories, Christchurch, New Zealand and the Department of Clinical Chemistry and Haematology, University Medical Centre, Groningen, Netherlands.