
Abstract
Familial hypercholesterolaemia (FH) is a common genetic disorder characterized by high plasma low-density lipoprotein (LDL)-cholesterol and premature coronary artery disease. Many factors, such as illness, high-dose statin therapy or a strict vegan diet can cause hypobetalipoproteinaemia (HBL). The more common secondary causes of HBL in the hospital setting include cachexia, intestinal malabsorption, malnutrition, severe liver disease and hyperthyroidism. We report a case of HBL in a 43-year-old man with previously demonstrated marked hypercholesterolaemia who attended a lipid disorders clinic for FH cascade screening. Surprisingly, a lipid profile taken at that time showed low plasma LDL-cholesterol and apolipoprotein B concentrations of 1.6 mmol/L and 0.61 g/L, respectively. He was not on lipid-lowering therapy. DNA sequencing showed that he was heterozygous for the LDLR gene mutation (C677R) present in other affected family members. Of interest, his serum transaminases were increased by ∼3-fold and hepatitis serology and genotyping confirmed a diagnosis of hepatitis C virus (HCV) infection. In summary, we describe a case of HBL secondary to chronic HCV infection in a patient with FH, confirmed by mutational analysis.
Introduction
Familial hypercholesterolaemia (FH) is a common autosomal codominant disorder caused by mutations in the low-density lipoprotein (LDL)-receptor (LDLR) gene that results in defective catabolism of LDL by the liver. 1,2 FH is characterized by high plasma LDL-cholesterol resulting in tissue cholesterol deposition and premature coronary artery disease. 3 Unfortunately, most people in Australia with FH are undiagnosed or diagnosed only after their first coronary event. 4 As such, family cascade screening has been advocated as a feasible, acceptable and cost-effective strategy to detect new FH cases. 5,6
Many factors, such as illness, high-dose statin therapy or a strict vegan diet, can cause hypobetalipoproteinaemia (HBL). 1,7,8 The more common secondary causes of HBL in the hospital setting include cachexia, intestinal malabsorption, malnutrition, severe liver disease and hyperthyroidism. Primary causes are rare and include abetalipoproteinaemia (ABL), chylomicron retention disease and familial HBL (FHBL). 1,7,8
Hepatitis C virus (HCV) is the principal viral cause of chronic hepatitis. 9 An association between chronic HCV infection and lipoprotein metabolism has been reported. 10 HCV can bind to lipoproteins 11 and has been shown to inhibit the microsomal triglyceride transfer protein (MTP), 12,13 a chaperone that facilitates apolipoprotein (apo) B-containing lipoprotein assembly and secretion. 14 Moreover, HCV-associated HBL has recently been shown to be negatively correlated with hepatic steatosis and HCV viral load. 15
In summary, we describe a case of HBL secondary to chronic HCV infection in a patient with FH, confirmed by mutational analysis.
Case report
A 43-year-old Caucasian male attended a lipid disorders clinic for FH cascade screening. The patient had been well, denied any cardiovascular symptoms and was on no medications. He was a smoker with a 30 pack-year history, consumed <10 g of alcohol per week and reported previous intravenous illicit drug use and time in prison. He undertook regular physical activity, and his diet was both healthy and balanced. A family history of FH (mother, two sisters and a brother) and premature coronary artery disease (father) was obtained (Figure 1a).
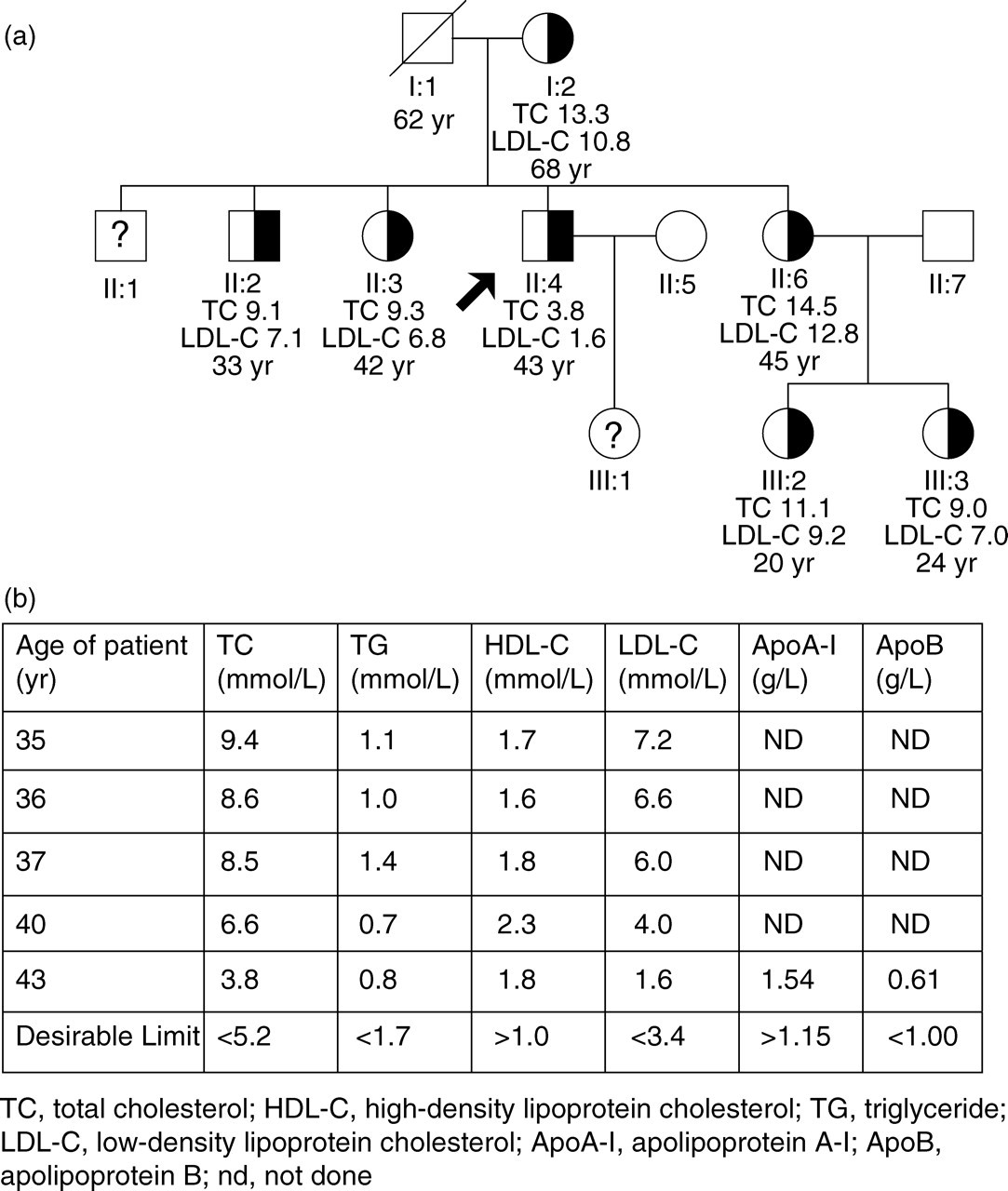
Pedigree and serial lipid results of the patient. (a) Pedigree of the family; arrow indicates the index patient. Black shading indicates LDLR C677R. Total cholesterol (TC) and LDL-cholesterol (LDL-C) concentrations in mmol/L are shown below each individual with age in years (yr). Question marks indicate no sample was able to be obtained for biochemical or genetic analysis. (b) Serial plasma lipid profiles of the patient off lipid-lowering treatment
On physical examination, he appeared well, with a body mass index of 23 kg/m2 (body weight 77.5 kg, height 1.83 m), pulse rate of 70 beats/min and blood pressure of 110/70 mmHg. His cardiorespiratory and gastrointestinal examinations were normal and he was clinically euthyroid. There were no peripheral manifestations of FH and, in particular, no evidence of Achilles tendon xanthomata or corneal arcus, in contrast to his mother (Achilles tendon xanthomata and corneal arcus) and affected siblings (Achilles tendon xanthomata only). Multiple skin tattoos were present.
Surprisingly, a lipid profile taken at that time showed low plasma LDL-cholesterol and apoB concentrations of 1.6 mmol/L and 0.61 g/L, respectively (<5th percentile for age and sex). In contrast, an lipid profile taken eight years earlier showed an LDL-cholesterol concentration of 7.2 mmol/L, consistent with heterozygous FH (Figure 1b). Serial lipid profiles demonstrated that his total cholesterol and LDL-cholesterol levels had gradually fallen by 60% and 78%, respectively, over that time period. He was not on lipid-lowering therapy.
DNA was extracted from peripheral blood leucocytes using standard Triton X-100 salting out procedure. Exon 14 of the LDLR was amplified by polymerase chain reaction (PCR) and sequenced using BigDye Terminator chemistry. The patient was found to be heterozygous for a Cys677Arg mutation (previously known as Cys656Arg) carried by other affected family members.
Liver function tests showed increased serum transaminase concentrations with alanine aminotransferase and aspartate aminotransferase activities of 127 and 100 U/L, respectively. Serum iron studies, copper, caeruloplasmin, and α1-antitrypsin were normal, and antimitochondrial antibody was negative. HIV1/2 antigen/antibody screening was negative. Antibody to HCV was detected by enzyme immunoassay screening and was confirmed positive by PCR for HCV RNA. He was found to be HCV genotype 3 with an HCV viral load of 1.04 × 10∧5 log10 (U/mL). There were no signs or symptoms of chronic liver disease. Abdominal ultrasonography showed normal liver size and echotexture, with no evidence of hepatic surface nodularity. He was referred to a hepatology clinic for consideration for antiviral treatment.
Discussion
We report the case of a patient with previously demonstrated marked hypercholesterolaemia consistent with the phenotypic manifestation of FH in his family who attended a lipid disorders clinic for FH cascade screening. Further laboratory investigations found that he had low-plasma LDL-cholesterol and apoB concentrations consistent with HBL, abnormal liver function tests and chronic hepatitis C. Subsequent genetic testing showed that he was heterozygous for the C677R LDLR mutation that was also present in his family members with FH. This missense mutation occurs within the epidermal growth factor precursor homology domain of the LDLR and is predicted to disrupt disulphide bond formation between Cys677 and Cys696. 16
HBL is characterized by <5th percentile levels of plasma LDL-cholesterol and apoB for age and sex. 2,7,8 Secondary causes of HBL include vegan diet, malnutrition, intestinal malabsorption, cachexia, hyperthyroidism, severe liver disease and high-dose statin treatment. Our patient was on a normal diet, showed no evidence of malabsorption or malnutrition, had normal thyroid function and was not on lipid-lowering therapy. However, liver function tests showed a ∼3-fold increase in serum transaminase concentrations and hepatitis serology and genotyping confirmed a diagnosis of HCV.
Hypocholesterolaemia in a patient with liver disease typically indicates advanced liver disease with poor prognosis. An association between chronic HCV infection and lipoprotein metabolism has been reported. 10 HBL secondary to HCV infection, particularly genotype 3, can occur in the absence of advanced liver disease and be corrected by antiviral treatment. 17 HCV can bind to a variety of lipoproteins. 11 Hepatic very-low-density lipoprotein (VLDL) assembly and secretion requires the association of apoB-100 and lipids, a process facilitated by MTP. 14 Of interest, hepatocyte HCV replication is dependent on this same process. 18
Although hepatic steatosis is observed in ∼50% of patients infected by HCV, liver ultrasonography was unremarkable in our patient, which may, in part, reflect the lack of sensitivity of this imaging modality. In a transgenic mouse model, over-expression of HCV core protein inhibited MTP activity and decreased hepatic VLDL secretion. 12 Any process that decreases the export of VLDL may cause steatosis. Indeed, hepatic steatosis has been reported in humans treated with MTP inhibitors, as well as in subjects with FHBL and ABL. 2,7,8 MTP inhibition has been implicated in HCV-related hepatic steatosis. 13 Moreover, HCV-associated HBL has recently been shown to be negatively correlated with hepatic steatosis and HCV viral load. 15 Taken together, these studies would be consistent with the concept that the inhibition of VLDL secretion by HCV core protein might induce liver steatosis and HBL.
In summary, we describe a case of HBL secondary to chronic HCV infection in a patient with FH, confirmed by mutational analysis. The lack of clinical manifestations of FH in this patient could relate to prolonged HCV-induced hypocholesterolaemia.